Durante el invierno de 1944, las tropas nazis cortaron todo el suministro de alimentos a los Países Bajos. Se calcula que murieron 20000 personas por inanición. Durante meses, la población tuvo que sobrevivir con una ingesta diaria muy pobre que finalizaría con la liberación del país y el fin de la II Guerra Mundial en mayo de 1945. Esta situación acarrearía terribles consecuencias para los bebés de las madres embarazadas en aquel momento.
Estudios han demostrado que los bebés gestados durante la hambruna tienen mayor tasa de enfermedades cardiovasculares, diabetes tipo 2, cáncer de mama, obesidad y esquizofrenia.
Al parecer, el entorno intrauterino de la madre mandó señales al feto para que silencie ciertos genes relacionados con el metabolismo y el desarrollo, como el gen Pim3. El mecanismo principal es mediante la metilación de ciertos genes, que conlleva a la represión transcripcional de estos. En concreto, se metila el carbono 5 de las citosinas de las islas CpG en los promotores de genes. Esto supone una señal para el reclutamiento de desacetilasas de histonas, lo que conlleva a la inactivación del gen. Entonces, estas modificaciones epigenéticas se mantendrían durante toda la vida, y prepararían al futuro individuo para un ambiente escaso de alimentos, en el que la prioridad es conservar y almacenar lo consumido, evitando el gasto. Esta teoría se conoce como el «fenotipo ahorrador» (David Barker, 1989). Por ejemplo, el gen Pim3 (proto-oncogén) implicado en la quema de energía, se expresa muy poco para no perder energía.
Teniendo en cuenta que estas personas luego se desarrollaron en un ambiente de «abundancia alimentaria», no es de extrañar que estas tiendan a acumular grasa, lo que se complica en mayor prevalencia de las patologías que hemos mencionado. Además, se ha visto que estas modificaciones epigenéticas se podrían transmitir a los nietos.
Por tanto, la situación de estrés puntual (hambruna), vivida en un momento tan crucial como es el inicio desarrollo, supone consecuencias para el resto de la vida (al igual que el ejemplo visto en clase de los ratones agoutí). Además, demuestra que la madre prepara al hijo para el futuro ambiente en el que va a crecer, incluso antes de nacer.
Por último, cabe destacar que aún queda mucho por investigar en este campo, pero se vuelve a poner de manifiesto la gran importancia de una buena alimentación y hábitos saludables, ya que tiene grandes repercusiones, tanto en nuestro bienestar físico como en la expresión del genoma de nuestros descendientes.
Institut Català d’Oncologia (ICO), Institut d’Investigació Biomèdica de Bellvitge (IDIBELL)
Estos datos permitirán a la comunidad científica generar hipótesis y priorizar sobre qué genes y variantes genéticas focalizar investigaciones futuras.
El estudio también ha permitido identificar variantes genéticas que modifican la expresión de los genes del colon y que están asociadas a enfermedades como el cáncer colorrectal, pero también con otros órganos como el cerebro, evidenciando la relación entre el intestino y el cerebro.
Un estudio liderado por investigadores del Programa de Análisis de Datos Oncológicos del Instituto Catalán de Oncología (ICO), el Grupo de Investigación en Cáncer Colorrectal del IDIBELL, la Universidad de Barcelona y el CIBER de Epidemiología i Salud Pública (CIBERESP) con la colaboración del grupo de investigación liderado por Grahan Casey, del Centro de Investigación en Salud Pública Genómica de la Universidad de Virginia (EE. UU.), ha puesto al alcance de la comunidad científica los resultados del análisis de más de 400 biopsias de colon sanos, creando así, el banco público más grande de datos sobre expresión de genes y variantes genéticas en el colon.
Los resultados del estudio se han publicado en un articulo científico en la revista Cellular and Molecular Gastroenterology and Hepatology liderado por el doctor Victor Moreno (ICO-IDIBELL-UB-CIBERresp).
Esquema de muestras y datos obtenidos en el estudio. Imagen: IDIBELL.
La identificación de los niveles de expresión de los genes en tejidos sanos es clave para entender por qué hay variantes genéticas asociadas a enfermedades, como el cáncer. En este sentido el proyecto, llamado ‘University of Barcelona and University of Virginia Genotyping and RNA Sequencing Project’ (BarcUVa-Seq), ha desarrollado una herramienta interactiva que pone a disposición pública los datos obtenidos, pudiendo así, explotar fácilmente la expresión genética del colon y las asociaciones con variantes genéticas obtenidas en el estudio. En este sentido la base de datos permitirá generar hipótesis, entender la función de marcadores genéticos ya asociados con el riesgo de sufrir una enfermedad de los que se conocen los mecanismos moleculares involucrados, y priorizar sobre que genes y variantes genéticas focalizar investigaciones futuras.
Asimismo, gracias a las biopsias analizadas han identificado variantes genéticas que regulan la expresión de genes involucrados en enfermedades del colon, como cáncer colorrectal o enfermedades inflamatorias, pero también de otros órganos como el cerebro, evidenciando la relación intestino-cerebro. Por ejemplo, se ha comprobado que parte de los marcadores genéticos asociados a la esquizofrenia estarían regulando genes en el colon. Además, en el estudio se proponen nuevos genes probablemente asociados con estas enfermedades.
Un proyecto innovador: BarcUVa-Seq
Este estudio nace de la colaboración con los investigadores de Estados Unidos con el objetivo principal de generar una base de datos epidemiológicos de referencia para entender la biología de las células epiteliales del colon (origen del cáncer colorrectal) y sus relaciones con factores genéticos y ambientales.
Concretamente BarcUVa-Seq, que es el nombre que recibe este proyecto, incluye datos de 485 voluntarios/as de la Región Metropolitana Sur, con pacientes reclutados en el Hospital Universitario de Bellvitge y el Hospital de Viladecans, sin ninguna lesión en el colon y recogidas en los últimos años durante una colonoscopia de cribado rutinario. De hecho, el tamaño muestral elevada y la homogeneidad en la recolección de las muestras ha supuesto un valor añadido, ya que no se conoce ningún otro estudio publicado con estas características hasta la fecha.
Referencia: Díez-Obrero V, Dampier CH, Moratalla-Navarro F, Devall M, Plummer SJ, Díez-Villanueva A, Peters U, Bien S, Huyghe JR, Kundaje A, Ibáñez-Sanz G, Guinó E, Obón-Santacana M, Carreras-Torres R, Casey G, Moreno V. Genetic Effects on Transcriptome Profiles in Colon Epithelium Provide Functional Insights for Genetic Risk Loci. Cell Mol Gastroenterol Hepatol. 2021 Feb 16:S2352-345X(21)00036-9.
Este artículo incluye una herramienta interactiva para comprender la información de una manera más visual. Se puede acceder a ella pinchando en «herramienta interactiva» que se encuentra en el párrafo siguiente a la imagen.
Un equipo de investigadores de la Universidad de Exeter, en Reino
Unido, ha elaborado un “reloj epigenético” para estimar la edad biológica del
cerebro. Este método, basado en las marcas epigenéticas del cerebro, puede ser
utilizado para estudiar cómo el envejecimiento acelerado del cerebro promueve
la aparición de enfermedades como el Alzhéimer o la demencia.
La edad biológica es un término que se utiliza para referirse al
estado funcional de cada uno de los órganos de un individuo. Mientras la edad
cronológica viene únicamente determinada por el tiempo vivido tras el
nacimiento, la edad biológica depende de múltiples factores, como el genoma del
individuo o su estilo de vida. En la actualidad existen algunos marcadores
moleculares que permiten conocer la edad biológica de un tejido, como la
longitud de los telómeros o algunas marcas epigenéticas.
El “reloj epigenético” obtenido por investigadores de la Universidad
de Exeter es una nueva herramienta para estimar la edad biológica del tejido
cerebral, en base a las marcas epigenéticas de su ADN. Los
resultados de esta investigación fueron publicados el pasado mes de octubre en
la revista Brain.
Para este estudio, los autores analizaron más de 1300 muestras de
tejido de la corteza cerebral de humanos de 1 a 108 años, en las que estudiaron
el perfil de metilación del ADN. El análisis determinó 347 marcadores
epigenéticos que permiten estimar la edad biológica del cerebro al ser
analizados conjuntamente.
Una vez determinados los marcadores epigenéticos, el equipo probó la
precisión de su reloj epigenético en más de 1200 muestras obtenidas de la
iniciativa Brains for Dementia Research. De igual modo, los
investigadores probaron el nuevo método en un conjunto de datos de 1175
muestras de sangre. En ambos casos, el test resultó efectivo para determinar la
edad biológica de las muestras.
La particularidad de este reloj molecular es que está dirigido a un
tejido concreto. “Nuestro estudio destaca la importancia de utilizar tejido que
sea relevante para el mecanismo que desea explorar al desarrollar modelos de
reloj epigenético. En este caso, el uso de tejido cerebral garantiza que el
reloj epigenético esté debidamente calibrado para investigar la demencia «,
explica la Dra. Gemma Shireby, investigadora en la Universidad de Exeter y
primera autora del trabajo.
Anteriormente ya se habían elaborado otros métodos basados en la
metilación del ADN para estimar la edad biológica, pero este nuevo reloj
molecular es todavía más preciso, lo que supone un gran avance. Tal y como explica
la Dra. Shireby, “Nuestro nuevo reloj epigenético ha superado drásticamente los
modelos anteriores en la predicción de la edad biológica en el cerebro humano.”
Ahora, el equipo de investigadores busca utilizar la nueva herramienta
para estimar la edad en cerebros de pacientes con Alzhéimer, donde esperan
obtener evidencias de envejecimiento acelerado. Esta aproximación puede ofrecer
datos esenciales para comprender mejor el envejecimiento cerebral y la
enfermedad de Alzheimer.
Investigadores del Instituto de Investigación contra la Leucemia Josep Carreras y el Instituto de Investigación Biomédica de Bellvitge han encontrado que los perfiles epigenéticos de una persona pueden influir en la gravedad de la enfermedad COVID-19. Los resultados del trabajo, publicado en EBioMedicine, abren el camino hacia el desarrollo de nuevos marcadores para predecir qué pacientes tienen un mayor riesgo a presentar una peor evolución de la enfermedad.
Los resultados del estudio, primero en considerar los factores epigenéticos en relación con la enfermedad COVID-19, indican que el epigenoma de las personas infectadas con el coronavirus SARS-CoV-2 influye en la gravedad de COVID-19. Imagen: Genotipia.
Desde el inicio de la pandemia de COVID-19 uno de los principales objetivos de la investigación biomédica ha sido determinar por qué unas personas son asintomáticas o presentan formas suaves de la enfermedad mientras que otras desarrollan formas más graves que requieren hospitalización o cuidados intensivos. Factores como tener una edad avanzada, ser hombre o presentar condiciones como la diabetes, la obesidad o la hipertensión contribuyen a definir la gravedad de la enfermedad COVID-19 en cada persona. Sin embargo, no son suficientes para realizar predicciones precisas para cada persona individual.
El trabajo dirigido por Manel Esteller, director del Instituto de Investigación contra la Leucemia Josep Carreras y Aurora Pujol, jefa del Grupo de Enfermedades Metabólicas del Instituto de Investigación Biomédica de Bellvitge, identifica factores moleculares que influyen en cómo va a responder cada persona a la infección por parte del coronavirus SARS-CoV-2.
Los investigadores se plantearon si además de los factores genéticos que empiezan a definirse en relación con la gravedad de COVID-19, podrían intervenir factores epigenéticos, mecanismos que regulan la expresión de los genes y la función del genoma sin alterar la secuencia de ADN.
Para identificar potenciales biomarcadores epigenéticos, el equipo analizó aproximadamente 850 000 posiciones del genoma susceptibles de ser metiladas, una de las marcas epigenéticas mejor caracterizadas, en muestras de sangre de 407 personas que habían resultado positivas para la presencia del coronavirus y no pertenecían a ningún grupo de riesgo (menores de 61 años sin trastornos autoinmunes, enfermedades pulmonares o cardiovasculares, hipertensión, diabetes u obesidad). A continuación, comparó los perfiles epigenéticos de aquellas personas que no mostraron síntomas, respecto a los de las personas que mostraron formas suaves de la enfermedad o necesitaron hospitalización y respiración asistida.
Los investigadores han encontrado 44 posiciones de metilación del genoma asociadas a la gravedad clínica de COVID-19. Entre ellas, 23 se encuentran localizadas cerca de 29 genes conocidos, que incluyen genes que intervienen en la respuesta al interferón, mecanismo implicado en la respuesta del sistema inmunitario a la exposición viral. “Encontramos que existían variaciones epigenéticas en los interruptores químicos que regulan la actividad del ADN en los positivos por el virus que desarrollaban un COVID-19 grave”, indica Manel Esteller. “Estas modificaciones suceden principalmente en genes asociados con una excesiva respuesta inflamatoria y en genes que reflejan una tendencia general a un peor estado de salud. Interesantemente, el 13{5f4e897c6ee23822ae50e890000b794346543cd330c4e8aa45664bf184d11e2e} de la población mundial presenta esta firma epigenética (EPICOVID), por tanto, es la población de máximo riesgo y a la que hemos de cuidar especialmente”, concluye el investigador.
Los investigadores han evaluado el potencial de la firma epigenética formada por el estado de metilación en las 44 posiciones del genoma para predecir la gravedad de la enfermedad en personas infectadas con el coronavirus y han encontrado que la firma EPICOVID, como ha sido denominado el perfil epigenético, muestra una elevada precisión (un 83.5{5f4e897c6ee23822ae50e890000b794346543cd330c4e8aa45664bf184d11e2e}) para predecir qué personas desarrollarán una forma grave de COVID-19.
Los resultados del estudio, primero en considerar los factores epigenéticos en relación con la enfermedad COVID-19, indican que el epigenoma de las personas infectadas con el coronavirus SARS-CoV-2 influye en la gravedad de COVID-19 e identifican un nuevo potencial biomarcador para estimar cómo afectará la enfermedad a una persona infectada con el coronavirus. Además, apoyan la importancia del papel de la respuesta inmunitaria frente al virus mediada por el interferón identificada en otros estudios.
La firma molecular epigenética podría tener importantes aplicaciones en el ámbito clínico. “Combinada con otros parámetros genéticos, celulares, serológicos y clínicos podría identificar pacientes que requieren de una monitorización precisa y tratamientos tempranos activos para prevenir la progresión de la enfermedad tanto como sea posible”, señalan los autores en el trabajo. “Con estas herramientas avanzadas de medicina personalizada, tanto del campo de la genómica como del epigenómica, es posible diseñar modelos predictivos que permitan detectar pacientes en riesgo de peor pronóstico y, por tanto, mejorar su tratamiento y evitar el colapso del sistema sanitario”, apunta Aurora Pujol. Además, en un escenario en el que la disponibilidad de vacunas no es absoluta, la firma EPICOVID podría contribuir a identificar a aquellas personas que podrían beneficiarse más de la vacunación.
Una investigación llevada a cabo en el Instituto de Neurociencias UMH-CSIC en Alicante, liderada por la Dra. Guillermina López-Bendito, demuestra por primera vez que es posible obtener neuronas específicas de una región cerebral determinada a partir de astrocitos, un tipo de células del sistema nervioso que llevan a cabo funciones muy importantes para el funcionamiento del cerebro.
Estos astrocitos han sido reprogramados mediante un factor proneural utilizando un virus para hacerle llegar a su destino en el cerebro de ratones. Además, con esta investigación han descubierto que los astrocitos expresan genes propios de sus neuronas “hermanas” (procedentes de una célula progenitora común), en cada región cerebral concreta, lo que ha hecho posible su reprogramación en un tipo de neurona sensorial específica.
“Hemos descubierto que genes clásicos de las neuronas también son expresados por los astrocitos, aunque en un nivel menor. Y que hay un código propio de cada región cerebral que comparten los astrocitos y las neuronas, y probablemente también otras células nerviosas. Esto es importante porque abre la posibilidad a recuperar en el futuro circuitos neuronales perdidos en ciegos o sordos congénitos”, explica la Dra. López-Bendito, que es directora de la Unidad de Neurobiología del Desarrollo del Instituto.
Este proyecto denominado “Reprogramación de células talámicas para el restablecimiento de circuitos sensoriales”, ha sido financiado por la Generalitat Valenciana con 400.000 euros y es la semilla para un nuevo proyecto impulsado por la Fundación La Caixa con 499.000 euros, a través de la Convocatoria CaixaResearch de Investigación en Salud.
Hasta hace poco los astrocitos se consideraban elementos secundarios en el cerebro. No obstante se ha demostrado su papel en diferentes funciones esenciales y en tareas que antes se consideraban exclusivas de las neuronas. Imagen de astrocitos y glía radial. Imagen: Jason Snyder CC BY 2.0).
Restaurar los sentidos perdidos
Gracias al tálamo, una estructura cerebral que actúa como un simulador del mundo exterior, antes del nacimiento el cerebro ya empieza la “puesta a punto” de sentidos como el tacto y la vista, como demostró el laboratorio de la doctora López-Bendito en trabajos anteriores.
Las dos estructuras cerebrales implicadas en este proceso son el tálamo, que recibe la información del exterior, y la corteza cerebral, que la procesa. Cuando hay una pérdida en la captación de los estímulos sensoriales, parte de las neuronas y los circuitos de estas dos regiones del cerebro se pierden o se reducen considerablemente.
Los astrocitos, un tipo de células nerviosas con forma de estrella, podrían ser cruciales para restaurar esos circuitos perdidos. Hasta hace poco se consideraba a estas células gliales “actrices secundarias” en el cerebro y la médula espinal. Consideradas tradicionalmente como proveedoras de alimento y soporte estructural a las neuronas, el papel de los astrocitos va más allá, y participan también en tareas que antes se consideraban exclusivas de las neuronas, como el procesamiento, la transferencia y el almacenamiento de información.
En esta línea, el descubrimiento del laboratorio de la Dra López-Bendito añade otra prueba del importante papel de los astrocitos, capaces de transformarse en neuronas al ser inducidos, con el potencial regenerativo que esto supone. Otro hallazgo de este trabajo es que las células que se generan en una zona concreta del cerebro, ya sean neuronas u otros tipos de células nerviosas, comparten una firma molecular. Es precisamente la expresión génica específica de cada región compartida con las neuronas, la que confiere a los astrocitos la capacidad de convertirse en neuronas de un tipo concreto en determinadas condiciones.
Células hermanas
La hipótesis de partida del grupo de Guillermina López-Bendito fue que, dado que las neuronas y los astrocitos se generan a partir de las mismas zonas germinales, podrían compartir firmas moleculares comunes que reflejen su origen y actúen potencialmente para coordinar las características de desarrollo específicas de la región a la que pertenecen.
Así han descubierto que los astrocitos del tálamo y de la corteza cerebral presentan firmas transcripcionales y epigenéticas específicas de la región a la que pertenecen. Esas firmas las comparten con las neuronas generadas dentro de la misma región, pero no con las de otras regiones. Y esas firmas compartidas entre ambos tipos de células proporcionan un grado notable de especificación regional para la reprogramación de astrocitos en neuronas inducida por un factor proneural, o gen maestro, denominado “Neurogenina 2”. Para introducir este gen maestro, el laboratorio de la Dra. López-Bendito ha inyectado en el tálamo postnatal de los ratones un virus que infecta solo a los astrocitos y logra reprogramarlos para que se conviertan en neuronas.
En otras palabras, como los astrocitos y las neuronas comparten progenitor y la expresión de genes específicos de una región, es esa firma molecular la que dirige la reprogramación inducida por factores de transcripción para que los astrocitos adquieran una identidad similar a la de sus neuronas hermanas.
Además, han visto que al manipular ese código genético específico de cada región se redirige la reprogramación de los astrocitos hacia neuronas de identidad regional diferente, pero predecible, en función de la manipulación efectuada.
Reparación espontánea
“Ahora estamos intentando averiguar si, de forma espontánea, los astrocitos pueden convertirse en neuronas en situaciones concretas. Por ejemplo, cuando provocamos un aumento de astrocitos reactivos”, explica la Dra. López-Bendito. Los astrocitos reactivos se encargan de proteger a las neuronas cuando se produce un daño, aunque en ocasiones su actuación también puede perjudicarlas si su reacción es muy potente.
El aumento del número de astrocitos reactivos, o astrogliosis, favorece que estas células se vuelvan más maleables o más “dóciles”. “En esas circunstancias pensamos que tal vez, sin necesidad de introducir un gen maestro que guíe la reprogramación, podríamos observar de forma espontánea esa capacidad de los astrocitos para convertirse en neuronas”, señala López-Bendito.
“Con este trabajo se demuestra que el proceso de reprogramación de astrocitos a neuronas es factible. Y lo hemos conseguido en estudios tanto in vitro como in vivo en ratones control. Ahora nuestro reto inmediato y proyecto presente es hacerlo posible en modelos de ratón con ceguera congénita. En estos animales utilizaremos esta misma técnica para reprogramar astrocitos sensoriales y que se conviertan en neuronas visuales que suplan a las que se habían perdido”, concluye la Dra López-Bendito.
El descubrimiento de una mosca blanca que utiliza un gen vegetal robado para eludir las defensas de su huésped puede ofrecer una ruta hacia nuevas estrategias de control de plagas
Este, es el primer ejemplo conocido de transferencia genética natural de una planta a un insecto. Explica una razón por la que la mosca blanca Bemisia tabaci es tan hábil para masticar cultivos: el gen que extrae de las plantas le permite neutralizar una toxina que algunas plantas producen para defenderse de los insectos.
Los primeros trabajos sugieren que la inhibición de este gen puede hacer que las moscas blancas sean vulnerables a la toxina, proporcionando una ruta potencial para combatir la plaga. Según Andrew Gloss, que estudia las interacciones planta-plaga en la Universidad de Chicago en Illinois "esto expone un mecanismo a través del cual podemos inclinar la balanza a favor de la planta ”. "Es un ejemplo notable de cómo el estudio de la evolución puede aportar nuevos enfoques para aplicaciones como la protección de cultivos".
La diminuta mosca blanca, que está más estrechamente relacionada con los pulgones que con las moscas, causa estragos en la agricultura en todo el mundo. Bemisia tabaci se encuentra entre las plagas de plantas más destructivas: beben savia azucarada de cientos de tipos de plantas, al tiempo que excretan una sustancia pegajosa y dulce llamada melaza que sirve como caldo de cultivo para el moho. Las moscas blancas también son vectores de más de 100 virus de plantas patógenos.
GENES ROBADOS
Que algunas especies de mosca blanca puedan deber parte de su habilidad depredadora a genes de otros organismos no es del todo sorprendente, porque el robo genético es común en la carrera armamentista entre las plantas y sus plagas. Durante millones de años, tanto las plantas como los insectos han tomado prestado en gran medida de los genomas microbianos, a veces utilizando sus genes recién adquiridos para desarrollar estrategias defensivas u ofensivas.
Algunos insectos, como el barrenador de los frutos del café (Hypothenemus hampei), han saqueado genes microbianos para extraer más nutrientes de las paredes celulares de las plantas difíciles de digerir, y un pariente silvestre del trigo ha robado un gen fúngico para combatir una enfermedad fúngica llamada Tizón de la cabeza. Pero hasta ahora no se sabía que las plantas y los insectos se robaran entre sí.
El entomólogo Youjun Zhang de la Academia China de Ciencias Agrícolas en Beijing y sus colegas estaban rastreando el genoma de B. tabaci en busca de genes robados, cuando encontraron uno que parecía haber evolucionado no en otros insectos o microbios, sino en plantas. Un estudio adicional mostró que el gen puede transferir un grupo químico a compuestos defensivos llamados glucósidos fenólicos. Muchas plantas, incluidos los tomates, fabrican estos compuestos para protegerse de las plagas, pero la modificación causada por el gen de la mosca blanca hizo que los compuestos fueran inofensivos.
Para probar la hipótesis, el equipo diseñó plantas de tomate para producir una molécula de ARN de doble hebra capaz de detener la expresión del gen de la mosca blanca. Casi todas las moscas blancas que posteriormente se alimentaron de estas plantas de tomate manipuladas murieron.
Ese resultado sugiere un nuevo medio de atacar a las moscas blancas, comenta Jonathan Gershenzon, ecologista químico del Instituto Max Planck de Ecología Química en Jena, Alemania. “Ofrece una enorme oportunidad de ser específico”, dice. "Podría mantener alejadas a las moscas blancas pero no dañar a los insectos beneficiosos como los polinizadores".
BATALLA DE PLANTAS Y PLAGAS
La transferencia de genes entre especies puede ser difícil de probar, dice el coautor del estudio Ted Turlings, ecólogo químico de la Universidad de Neuchâtel en Suiza. Para hacerlo, Zhang, Turlings y sus colegas analizaron las secuencias de genes similares en plantas y determinaron que el gen de la mosca blanca era su pariente evolutivo. El equipo también llevó a cabo análisis para demostrar que el gen estaba integrado en el genoma de la mosca blanca y no era el resultado de muestras contaminantes del ADN de las plantas.
Los resultados fueron sorprendentes, pero convincentes, dice Yannick Pauchet, entomólogo molecular que también trabaja en el Instituto Max Planck de Ecología Química. “Según los datos que proporcionan, la transferencia horizontal de genes es la explicación más convincente”
Pero no está claro cómo logró la mosca blanca deslizar un gen de una planta. Una posibilidad, según Turlings, es que un virus sirviera como intermediario, transportando material genético de una planta al genoma de la mosca blanca. A medida que los investigadores secuencian más genomas, es posible que descubran más ejemplos de transferencia de genes entre plantas y animales, dice Gloss.
"Los insectos que toman los genes de las plantas son solo la última parte del arsenal que aún no habíamos encontrado", comenta. "En la batalla entre las plantas y sus plagas de insectos o patógenos, se extraen genes de todo el árbol de la vida".
Pongo el enlace: https://www.nature.com/articles/d41586-021-00782-w?s=09
Una investigación realizada en el CNIC y Hospital Universitario Virgen de Arrixaca de Murcia demuestra que la hematopoyesis clonal representa un nuevo factor de riesgo cardiovascular y un vínculo relevante entre edad y enfermedad cardiovascular
Una persona adulta genera diariamente cientos de miles de millones de células sanguíneas. Sin embargo, este proceso necesario facilita la aparición de mutaciones en las células responsables de su producción. Dichas mutaciones se denominan somáticas porque se producen debido a cambios adquiridos, no hereditarios, en el ADN de estas células progenitoras. Y, aunque estas mutaciones son en su mayor parte inocuas, en ocasiones provocan que las células que las presentan adquieran una ventaja competitiva que permite que se expandan de forma progresiva, dando lugar a clones en la sangre, lo que conocemos como hematopoyesis clonal.
Ahora, un equipo de investigadores del Centro Nacional de Investigaciones Cardiovasculares (CNIC) y del Hospital Universitario Virgen de Arrixaca de Murcia describe que la presencia de estas mutaciones adquiridas en las células sanguíneas indica un riesgo elevado de progresión acelerada de insuficiencia cardiaca, una de las causas más importantes de mortalidad en el mundo.
La hematopoyesis clonal está relacionada con el envejecimiento, por lo que a medida que envejecemos hay más probabilidades de que se produzca este proceso, explica el Dr. José Javier Fuster, coordinador de la investigación que se publica en The Journal of the American College of Cardiology (JACC).
“Estudios recientes han demostrado que los individuos con hematopoyesis clonal tienen un mayor riesgo de desarrollar cánceres hematológicos y de muerte, que, sin embargo, no está causada por la enfermedad oncológica, sino por causas cardiovasculares”, asegura el Dr. Fuster.
Este nuevo conocimiento ha generado un gran interés por evaluar la posibilidad de que la hematopoyesis clonal contribuya al mayor riesgo de enfermedad cardiovascular asociado al envejecimiento.
No hay que olvidar que la insuficiencia cardiaca es la principal causa de hospitalización en mayores de 65 años y una importante causa de morbimortalidad.
“Sabemos que existe una relación clara entre la hematopoyesis clonal y un mayor riesgo de desarrollar enfermedad cardiovascular aterosclerótica, la causa última de la mayoría de infartos de miocardio y de muchos ictus cerebrales”, indica el Dr. Domingo Pascual-Figal, investigador externo del CNIC y cardiólogo del Hospital Universitario Virgen de Arrixaca de Murcia.
El estudio muestra la importancia de la hematopoyesis clonal como proceso patogénico que acelera y agrava la progresión de la insuficiencia cardiaca
Además, trabajos experimentales previos llevados a cabo por investigadores del CNIC han demostrado que ciertas mutaciones que conducen a la hematopoyesis clonal aceleran el desarrollo de la aterosclerosis y a la progresión de la insuficiencia cardíaca en ratones.
Investigadores del CNIC que han participado en el estudio: Mª Ángeles Zuriaga, Mirian Díez, Fátima Sánchez-Cabo, Ana Dopazo, José Javier Fuster, Ana Quintas y Jorge de la Barrera. Imagen: CNIC.
En este nuevo estudio, en el que también han participado las Unidades de Genómica y Bioinformática del CNIC e investigadores del Hospital Universitario Germans Trias i Pujol de Badalona, se ha analizado cómo la presencia de mutaciones ligadas a hematopoyesis clonal afecta a la evolución clínica de pacientes con insuficiencia cardíaca, tanto isquémica como con otros orígenes.
Los investigadores secuenciaron el ADN genómico de células sanguíneas de un grupo de pacientes diagnosticados con insuficiencia cardíaca que fueron monitorizados durante años con el fin de detectar la presencia de hematopoyesis clonal y valorar su posible conexión con la evolución de esta enfermedad.
Los resultados, indica el Dr. Fuster, apuntan que, independientemente del origen de la insuficiencia cardíaca, “la presencia de estos clones mutantes en sangre agrava la progresión de la insuficiencia cardiaca y empeora su pronóstico”. En concreto, detalla el Dr. Pascual-Figal , “los clones con mutaciones en dos genes frecuentemente ligados a hematopoyesis clonal, TET2 y DNMT3A, se asociaron con un mayor riesgo de hospitalizaciones y muertes debidas a la propia insuficiencia cardiaca”.
Para los investigadores, estos datos “ponen de manifiesto la importancia de la hematopoyesis clonal como proceso patogénico que acelera y agrava la progresión de la insuficiencia cardiaca, independientemente de la presencia de enfermedad aterosclerótica”.
Los autores concluyen que su estudio apoya la idea emergente de que la “hematopoyesis clonal representa un nuevo factor de riesgo cardiovascular y un vínculo relevante entre edad y enfermedad cardiovascular”. Además, aseguran, “abren la puerta al desarrollo de terapias personalizadas dirigidas a aquellos pacientes que presentan estas mutaciones somáticas y, con ello, prevenir la progresión de patologías muy relevantes como la insuficiencia cardiaca”.
El CNIC está desarrollando diversos proyectos para profundizar en los efectos de las mutaciones somáticas y la hematopoyesis clonal, con el objetivo de experimentar estrategias de prevención y tratamiento personalizadas para los pacientes que desarrollan este tipo de mutaciones.
La investigación ha sido financiada por la Beca Leonardo para Investigadores y Creadores Culturales 2019 de la Fundación BBVA, el Instituto de Salud Carlos III, el Ministerio de Ciencia e Innovación y la Fundación Séneca de Ciencia y Tecnología de la Región de Murcia. José Javier Fuster forma parte de la Red «Clonal hematopoiesis and atherosclerosis» financiada por la Fundación Leducq.
Referencia: Pascual-Figal, D. A., et al. Clonal Hematopoiesis and Risk of Progression of Heart Failure With Reduced Left Ventricular Ejection Fraction. Journal of the American College of Cardiology. 2021. 77(14), 1747-1759. https://www.jacc.org/doi/10.1016/j.jacc.2021.02.028
Investigadores de la Universidad de California San Diego han desarrollado una estrategia de terapia génica que permite regular la tolerancia al dolor a través del control de un gen implicado en la percepción del dolor. El equipo ha testado la aproximación en diferentes modelos en ratón, donde han detectado una menor sensibilidad al dolor de forma duradera. Los resultados se han publicado en Science Translational Medicine.
Una de cada 5 personas adultas sufre dolor crónico, que afecta en mayor o menor medida a su actividad cotidiana y puede comprometer seriamente su calidad de vida. Si bien existen algunas estrategias terapéuticas para tratar el dolor crónico, éstas conllevan inconvenientes, como el riesgo a desarrollar adicción en el caso de los opiodes. En estas circunstancias, cada vez es más necesario obtener nuevos tratamientos dirigidos a manejar el dolor.
La aproximación para tratar el dolor crónico que ha desarrollado el equipo de la Universidad de California San Diego parte de la identificación, en un estudio previo, de una mutación en el gen que codifica para la proteína NaV1.7. La presencia de dos copias dañadas del gen lleva a que las personas portadoras no sientan dolor, por lo que los investigadores se plantearon si la inactivación temporal del gen podía representar una opción terapéutica para el dolor crónico.
Los investigadores han desarrollado una estrategia de terapia génica que no modifica el ADN sino que modula la expresión de un gen. Imagen: Genotipia.
Los investigadores han utilizado dos plataformas moleculares diferentes para inactivar la expresión del gen que codifica NaV1.7 de forma no permanente. En primer lugar, han utilizado un sistema basado en herramientas CRISPR, conocidas por su capacidad para introducir cambios en el material hereditario. En este caso, el sistema CRISPR no está dirigido a modificar la secuencia de ADN, como es lo habitual, sino a regular los mecanismos epigenéticos que median la expresión del gen. Para ello, en lugar de utilizar la proteína Cas9 funcional que corta el ADN el equipo ha utilizado una enzima Cas9 inactiva, acoplada a ARN guía que se une al gen que codifica NaV1.7 y bloquea su expresión.
“No elimina ningún gen, por lo que no hay cambios permanentes en el genoma”, señala Ana Moreno, investigadora en la Escuela de Ingeniería de la Universidad de California San Diego y primera firmante del trabajo. “Una de las principales preocupaciones con la edición genética mediante CRISPR son los efectos en otras regiones del genoma. Una vez se corta el ADN no se puede ir atrás. Con Cas9 inactiva, no hacemos nada irreversible”.
En segundo lugar, los investigadores utilizaron una plataforma basada en proteínas sintéticas con dedos de zinc. Esta aproximación es más compleja a nivel técnico, pero presenta como ventaja que al generarse proteínas que no existen en la naturaleza, es menos probable que el organismo haya desarrollado una respuesta inmunitaria frente a ellas. En el caso de CRISPR, puesto que las proteínas Cas utilizadas derivan de bacterias relativamente comunes, el riesgo a que el sistema inmunitario las reconozca y actúe frente a ellas es mayor.
El equipo evaluó y optimizó la efectividad de ambas plataformas moleculares para reprimir la expresión del gen que codifica NaV1.7. en cultivos celulares y a continuación evaluó su eficacia frente al dolor en diferentes modelos de dolor en ratón. Para ello empaquetaron los componentes del sistema CRISPR o de las proteínas con dedos de zinc en vectores víricos que administraron directamente en el sistema nervioso de los animales.
Tras el tratamiento, los investigadores observaron que disminuía la sensibilidad a los diferentes tipos de dolor (inflamatorio o inducido por quimioterapia) de los animales. Esta efectividad en la terapia se mantenía tras 44 semanas en el caso del dolor inflamatorio y 15 semanas en el dolor inducido por quimioterapia. Además, los animales tratados no mostraron cambios en la función motora.
Los resultados preliminares en los modelos de ratón indican que la analgesia de larga duración inducida vía represión epigenética (denominada LATER, por sus siglas en inglés), podría ser una estrategia muy prometedora para el tratamiento de diferentes tipos de dolor crónico mediados por un aumento en la actividad de Nav1.7. Este es el caso de la polineuropatía diabética, la osteoartritis y la eritromelalgia, entre otros. Además, también podría resultar beneficiosa para los pacientes con dolor asociado a tratamientos por quimioterapia.
Una de las ventajas de esta estrategia es que no se elimina de forma permanente la capacidad para sentir dolor por lo que podría utilizarse para tratar condiciones en las que el dolor se mantiene a plazo largo pero no es permanente. “Pensamos en el joven atleta o en un soldado herido en la guerra en los que el dolor se puede resolver con la curación de las heridas”, indica Tony Yaksh, profesor de Anestesiología y Farmacología en la Universidad de California San Diego y uno de los investigadores seniors del estudio. “No querríamos eliminar de forma permanente la capacidad para sentir dolor en estas personas, especialmente si tienen una esperanza de vida elevada. Esta aproximación CRISPR/Cas9 inactiva ofrece a esta población una intervención terapéutica alternativa, lo que es un gran paso en el campo del manejo del dolor”.
De momento, el siguiente paso de los investigadores será optimizar los protocolos e iniciar pruebas de toxicidad en modelos con primates no humanos. Además, para favorecer la traslación de la estrategia LATER a la práctica clínica, los investigadores han creado la empresa Navega Therapeutics.
Referencia: Moreno AM, et al. Long-lasting analgesia via targeted in situ repression of NaV1.7 in mice. Sci Transl Med. 2021 Mar 10;13(584) doi: http://dx.doi.org/10.1126/scitranslmed.aay9056
Ha sido localizada en el estado occidental de Maharashtra tras varias pruebas en la secuenciación del genoma del virus
La India informó este miércoles de la existencia de una «variante doble mutante» del SARS-CoV-2, además de otras cepas importadas, en estudios realizados en varios estados del país, en medio de una segunda ola de contagios que sigue en aumento, con 47.262 nuevos positivos y 275 fallecidos en las últimas 24 horas.
El Consorcio indio sobre genómica del SARS-CoV-2 (INSACOG), una agrupación de diez laboratorios nacionales formada por el Ministerio de Salud y Bienestar Familiar, confirmó que esta nueva mutación se detectó, sobre todo, en el estado occidental de Maharashtra tras varias pruebas en la secuenciación del genoma del virus.
«El análisis de muestras de Maharashtra ha revelado que, en comparación con diciembre de 2020, ha habido un aumento en la fracción de muestras con las mutaciones E484Q y L452R«, explica el informe, que añade que «estas mutaciones se han encontrado en aproximadamente el 15-20{5f4e897c6ee23822ae50e890000b794346543cd330c4e8aa45664bf184d11e2e} de las muestras y no coinciden con ningún COV previamente catalogado«
La india es el tercer país del mundo con mayor número de casos de coronavirus y de decesos del mundo, según los datos recabados por la Universidad Johns Hopkins. A día de hoy suma 11.734.058 de casos detectados y 160.441 decesos.
Secuenciación del virus
En el estado meridional de Kerala se han secuenciado 2.032 muestras de la variante N440K, que se detectó previamente en otros 16 países, incluidos el Reino Unido, Dinamarca, Singapur, Japón y Australia.
Esta variante del SARS-CoV-2 se encontró además en el 33 {5f4e897c6ee23822ae50e890000b794346543cd330c4e8aa45664bf184d11e2e} de las muestras analizadas en el estado de Andhra Pradesh y en 53 de las 104 pruebas de Telangana, ambos situados en el sur de la India.
También detectaron en las 10.787 pruebas analizadas por el INACOG, 736 casos de cepa británica, 34 personas contagiadas de la variante sudafricana y una muestra de la variante brasileña.
Sin embargo, el reporte subraya que «aunque se hayan encontrado varias cepas preocupantes y una nueva variante doble mutante en la India, no se han detectado en cantidades suficientes para establecer o relacionar el rápido aumento de casos en algunos estados«.
Las autoridades advirtieron que estas nuevas variantes requieren la misma respuesta epidemiológica y de salud pública que otras cepas detectadas en otros países, como «el seguimiento de contactos estrechos, el aislamiento inmediato de positivos, así como el seguimiento del Protocolo Nacional que establezca cada región» del país asiático.
El anuncio de la nueva variante se produce en plena segunda ola de contagios en la India, con 47.262 nuevos casos registrados en las últimas 24 horas, un contraste con el momento de optimismo vivido el mes pasado, cuando se detectaron menos de 10.000 positivos diarios.
La ataxia de Friedreich es una enfermedad hereditaria neurodegenerativa con una prevalencia estimada en España de 4,7 por cada 100000 habitantes. Está causada principalmente por mutaciones de expansión del triplete GAA en el primer intrón del gen FXN, lo cual disminuye la expresión de la proteína frataxina. Debido a ello, la enfermedad implica una serie de manifestaciones neuromusculares y cardíacas en los pacientes, que incluyen una pérdida progresiva de la capacidad de movimiento y posicionamiento y dificultad para hablar y tragar, además del desarrollo de diabetes mellitus. Actualmente, no existe una cura o tratamiento eficaz para la enfermedad.
Existen dos estructuras celulares principalmente encargadas de mantener una correcta regulación de los niveles de calcio que circulan por la célula: el retículo endoplásmico y las mitocondrias. Imagen: Representación de una célula animal típica, en la que se observa el retículo endoplásmico y mitocondrias. Judith Stoffer, National (Institute of General Medical Sciences, NIGMS).
Para que las células funcionen correctamente, es necesario que se mantenga una comunicación coordinada entre las diferentes estructuras y procesos celulares. Un ejemplo de ello es la homeostasis del calcio celular. El calcio es un catión que actúa como mensajero en la célula, mediando en la regulación de procesos esenciales como la producción de energía y la muerte celular. Existen dos estructuras celulares principalmente encargadas de mantener una correcta regulación de los niveles de calcio que circulan por la célula y que, por tanto, regulan todos estos procesos. Por un lado, el retículo endoplasmático es el encargado de almacenar el calcio y cuando se da la señal apropiada, éste es liberado hacia el citoplasma. El compartimento encargado de recoger ese calcio liberado es la mitocondria. El intercambio de calcio entre los compartimentos es muy rápido, por lo que ambas estructuras deben coordinarse a través de puentes proteicos que se establecen entre proteínas localizadas en sus membranas. Estas estructuras se denominan membranas asociadas a retículo endoplasmático y mitocondria (del inglés Endoplasmic reticulum-mitochondria associated membranes o MAMs) y es común que presenten alteraciones en su función o estructura en enfermedades neurodegenerativas como el Huntington o la Esclerosis Lateral Amiotrófica.
Desde el punto de vista molecular, la ataxia de Friedreich presenta una disfunción en procesos relacionados con la mitocondria, que incluyen un desequilibrio oxidativo y una menor producción energética celular. A pesar de que en estudios anteriores se habían sugerido alteraciones en la regulación del calcio en varios modelos de la enfermedad, nuestro grupo ha demostrado el mecanismo por el cuál se da esta alteración.
Utilizando como modelo una línea celular de neuroblastoma, hemos observado que las células deficientes en frataxina presentan alteraciones a nivel estructural y funcional en las MAMs. Los puentes de comunicación entre ambos compartimentos se encuentran disminuidos, impidiendo que la mitocondria capte correctamente el calcio procedente del retículo endoplasmático. El tratamiento con dos antioxidantes, un derivado de la vitamina E y N-acetil cisteína, es capaz de revertir estos defectos, poniendo de manifiesto la importancia del efecto del estrés oxidativo en estas estructuras.
Asimismo, hemos observado la localización de frataxina en el dominio de las MAMs y su interacción con proteínas esenciales en el intercambio de calcio, algo que no se había descrito hasta el momento. Esto sugiere una nueva función de la proteína en la que podría estar involucrada en la regulación de estas estructuras.
Por otra parte, en el estudio también se ha utilizado Drosophila melanogaster como modelo de la enfermedad. Tras promover la entrada de calcio a la mitocondria de forma directa, se han podido recuperar algunos defectos provocados por la deficiencia de frataxina en la mosca, como la habilidad motora, la supervivencia y la producción de energía celular.
En definitiva, los resultados indican que el deterioro en el proceso de intercambio de calcio es un factor principal en la patología de la ataxia de Friedreich, aportando una nueva posible función de la frataxina no descrita hasta el momento y desplegando un nuevo campo de investigación en el análisis de las MAMs como vías terapéuticas de la enfermedad.
Artículo de referencia: Rodríguez LR et al. Oxidative stress modulates rearrangement of endoplasmic reticulum-mitochondria contacts and calcium dysregulation in a Friedreich’s ataxia model. Redox Biol. 2020 Oct;37:101762. doi: http://dx.doi.org/10.1016/j.redox.2020.101762
PUBLICADO EL ABRIL 1, 2021 Amparo Tolosa, Genotipia
El primer análisis genómico detallado de la placenta humana confirma que este tejido contiene múltiples anomalías genéticas y revela la presencia de patrones de mutaciones encontrados de forma común en cánceres pediátricos. El estudio, dirigido por el Wellcome Sanger Institute y la Universidad de Cambridge y publicado recientemente en Nature, ofrece por primera vez la posibilidad de conocer los cambios genéticos que se producen en las poblaciones celulares de la placenta y evaluar el potencial impacto de las mutaciones en el embarazo o el desarrollo embrionario.
La placenta es un órgano esencial para el desarrollo embrionario de mamíferos. Conecta el feto en desarrollo a las paredes del útero materno, proporciona oxígeno y nutrientes y libera hormonas importantes para el embarazo. Pese a su relevancia, a diferencia de otros órganos y tejidos, hasta el momento no se había analizado en profundidad su arquitectura genética. El estudio dirigido por el Wellcome Sanger Institute y la Universidad de Cambridge ha abordado por primera vez esta cuestión con resultados muy interesantes.
Los investigadores han analizado 86 biopsias y 106 microdisecciones de distintas regiones de la placenta de 42 embarazos humanos diferentes y han comparado su composición genética con el ADN materno y el derivado del cordón umbilical para identificar tanto las mutaciones ocurridas en la placenta como la secuencia en la que pudieron producirse.
La placenta es un órgano de «parches» genéticos
Los resultados del trabajo muestran a la placenta como un tejido formado por “parches” de poblaciones de diferente composición genética, que los investigadores identifican como expansiones clonales, poblaciones celulares derivadas de una misma célula ancestral. Los investigadores también destacan la presencia de una tasa mutacional mayor que en otros tejidos, firmas mutacionales únicas en tejidos no tumorales y frecuentes cambios en el número de copias de fragmentos de ADN. “Nuestro estudio confirma por primera vez que la placenta está organizada de forma diferente a cualquier otro órgano humano y que, de hecho, se parece a una tela de retales de tumores”, señala Steve Charnock-Jones, investigador de la Universidad de Cambridge y uno de los directores del trabajo. “La tasa y los patrones de mutaciones genéticas eran también increíblemente altos en comparación con otros tejidos humanos sanos”.
Interesantemente, algunos de los perfiles de mutaciones presentes en las células de la placenta son comunes en tumores pediátricos, lo que apunta a semejanzas en los mecanismos de formación de la placenta y dichos tumores. No obstante, a diferencia del tejido canceroso la evolución clonal de las poblaciones de la placenta no parece estar dirigida por mutaciones concretas.
Dentro de las posibles causas de la característica arquitectura genética de la placenta los investigadores apuntan a diferencias entre tejidos adultos y fetales, así como al carácter temporal de la placenta, que puede contribuir a que los mecanismos para proteger el material hereditario no sean los mismos que en otros tejidos.
Los resultados del trabajo muestran a la placenta como un tejido formado por “parches” de poblaciones de diferente composición genética. Imagen: Rosario García, Genotipia.
La arquitectura genética de la placenta deriva de cuellos de botella poblacionales
A partir de la comparación de la composición genética de las muestras de placenta y de cordón umbilical los investigadores han reconstruido parcialmente la historia de cambios genéticos ocurridos en la placenta de cada embarazo evaluado. Mediante esta aproximación han estimado que los linajes celulares de la placenta se separan genéticamente de los que darán lugar al embrión y otras estructuras extraembrionarias a través de cuellos de botella poblacionales. “Estos cuellos de botella podrían representar rutas del desarrollo a través de las cuales las células anormales citogenéticamente se separan espacial y filogenéticamente”, señalan los autores del trabajo. Esta situación podría representar una oportunidad para que se normalice el contenido cromosómico en embriones derivados de zigotos con aneuploidías o número alterado de cromosomas.
El equipo pone como ejemplo de estos cuellos de botella un caso de rescate cromosómico identificado en el estudio. Los investigadores detectaron la presencia de una trisomía del cromosoma 10 en una de las muestras de una placenta. No obstante, en otras regiones de este órgano y en las células del cordón umbilical se observaban únicamente dos copias del cromosoma 10, ambas maternas.
A partir del análisis genético el equipo estima que, en el momento de la fecundación, el óvulo tenía dos copias diferentes del cromosoma 10, que sumadas al cromosoma 10 paterno presente en el espermatozoide daban lugar a una trisomía. Sin embargo, en las primeras divisiones tras la fecundación, una de las células, de la que derivarían posteriormente el embrión y parte de la placenta, perdió el cromosoma paterno y revirtió la trisomía.
“Fue fascinante observar cómo un defecto genético tan grave como un error en el número de copias de cromosomas era limado por el bebé, pero no por la placenta”, destaca Gordon Smith, investigador de la Universidad de Cambridge y uno de los directores del trabajo. “Este error debía estar presente en el óvulo fecundado y sin embargo las poblaciones de células derivadas y, lo que es más importante, las que iban a formar el bebé, tenían el número correcto de copias del cromosoma 10, mientras que partes de la placenta fallaron en hacer esa corrección”.
Los investigadores detectaron la presencia de una trisomía del cromosoma 10 en una de las muestras de una placenta. No obstante, en otras regiones de este órgano y en las células del cordón umbilical se observaban únicamente dos copias del cromosoma 10, ambas maternas. Imagen: Rosario García, Genotipia.
Una base para estudiar el efecto de las alteraciones genéticas de la placenta en el embarazo
Los investigadores describen la placenta como un tejido heterogéneo a nivel genético, lo que explica las dificultades que han tenido estudios previos a la hora de evaluar los efectos de las alteraciones genéticas de la placenta en la función de este órgano y en la aparición de complicaciones durante el embarazo. Se sabía que en algunos embarazos algunas células de la placenta mostraban alteraciones genéticas no presentes en el feto pero no se había investigado en detalle la estructura genética del órgano.
El tamaño de la muestra no ha permitido que el equipo de investigadores pudiera relacionar alteraciones genéticas de la placenta con el desenlace de los embarazos estudiados. Investigaciones futuras deberán abordar esta cuestión, así como la contribución de las alteraciones del genoma de las poblaciones celulares de la placenta en la función de este órgano que puedan llevar a complicaciones obstétricas como la preeclampsia o el parto prematura.
“La placenta es semejante al salvaje oeste del genoma humano, completamente diferente en su estructura a cualquier otro tejido humano sano”, señala Sam Behjati, investigador en el Wellcome Sanger Institute y uno de los directores del trabajo. “Nos ayuda a protegernos de defectos en nuestro código genético, pero permanece como una carga elevada de enfermedad asociada a la placenta. Nuestros resultados proporcionan una base sobre la que estudiar la asociación entre las aberraciones genéticas en la placenta y los resultados en el nacimiento, a gran resolución y a escala masiva”.
Investigadores del Memorial Sloan Kettering Cancer Center defienden la utilidad clínica de secuenciar el genoma germinal de todos los pacientes pediátricos con tumores sólidos y estimar su predisposición genética al cáncer en un reciente artículo publicado en Nature Medicine.
Los investigadores han comparado el genoma germinal de 751 pacientes pediátricos con tumores sólidos con el genoma correspondiente a sus tumores y el de sus progenitores, para estimar su predisposición genética al cáncer.
La baja frecuencia con la que se presenta el cáncer en niños ha llevado a que sus causas y mecanismos no se conozcan de forma completa y a que parte de los responsables genéticos, hereditarios o no, sigan siendo desconocidos. En la actualidad, se estima que aproximadamente uno de cada 10 pacientes pediátricos de cáncer presentaba una predisposición genética hereditaria elevada a la enfermedad y diversos estudios han identificado algunas mutaciones en genes concretos que alertan de esta predisposición. Sin embargo, la comprensión incompleta de las bases genéticas y hereditarias del cáncer pediátrico puede influir en los criterios que deben cumplir los pacientes para realizarles pruebas genéticas y repercutir en el manejo de la enfermedad o la identificación de otros miembros de la familia potencialmente afectados.
Un equipo de investigadores del Memorial Sloan Kettering Cancer Center ha realizado el mayor estudio de mutaciones germinales de predisposición al cáncer pediátrico hasta la fecha cuyos resultados refuerzan la utilidad clínica de este tipo de análisis tanto para los pacientes como para sus familias.
Los investigadores analizaron el genoma del cáncer y el genoma somático de 751 pacientes pediátricos con tumores sólidos para identificar la posible presencia de variantes germinales, presentes en todas las células y, por tanto, heredables. Para ello utilizaron un ensayo de secuenciación masiva desarrollado por el equipo, MSK-IMPACTTM, capaz de identificar cambios en la secuencia, alteraciones en el número de copias y otras alteraciones genéticas en 468 genes diferentes.
El equipo identificó una o más variantes germinales patogénicas o probablemente patogénicas en un 18{5f4e897c6ee23822ae50e890000b794346543cd330c4e8aa45664bf184d11e2e} de los pacientes. Esta proporción se reducía a un 13{5f4e897c6ee23822ae50e890000b794346543cd330c4e8aa45664bf184d11e2e} si se consideraba únicamente variantes en genes dominantes con media o alta penetrancia.
Interesantemente, un número elevado de las variantes identificadas no se correspondía con lo esperado según las características del paciente, lo que cuestiona los criterios que se utilizan en la actualidad para decidir si se realiza un análisis de variantes germinales en un paciente pediátrico. “En cerca de la mitad de los pacientes donde hemos encontrado una predisposición hereditaria, no habríamos predicho la detección de una mutación de predisposición al cáncer y no la habríamos rastreado,” señala Michael Walsh, director de investigación en genómica del cáncer pediátrico en el Centro de Genómica del Cáncer Hereditario del Memorial Sloan Kettering Cancer Center y director del estudio.
Los pacientes en los que se identificó un resultado positivo para variantes de predisposición fueron derivados al Servicio de Genética Clínica del centro, donde se investigó en detalle si habían recibido la mutación de sus progenitores o si había otros miembros de la familia portadores de la variante en cuestión.
Los resultados del trabajo aportan nueva información sobre el desarrollo del cáncer en niños: confirman la participación de algunas variantes y genes y amplían la colección de mutaciones que aumentan la predisposición al cáncer. Además, plantean cuestiones importantes sobre en qué casos realizar el cribado de mutaciones germinales.
Los investigadores defienden el análisis de mutaciones germinales de todos los pacientes pediátricos con cáncer, así como el ofrecimiento de asesoramiento genético a los familiares. Como razones para utilizar esta estrategia destacan la posibilidad de guiar el tratamiento de forma más precisa, identificar otros familiares potencialmente afectados o estimar el riesgo de tener otro hijo afectado. Además, la identificación de variantes de predisposición elevada al cáncer puede ser relevante también cuando los pacientes alcanzan la edad adulta y piensan en tener descendencia.
Referencia: Fiala, EM, et al. Prospective pan-cancer germline testing using MSK-IMPACT informs clinical translation in 751 patients with pediatric solid tumors. Nat Cancer 2, 357–365 2021. https://doi.org/10.1038/s43018-021-00172-1
Un estudio evolutivo realizado por investigadores de la Universidad de Búfalo y la Universidad de Chicago ha relacionado un gen, HAND2, con el inicio del parto en humanos. El trabajo sugiere que este gen podría estar relacionado también con otros procesos relevantes de la gestación, como la comunicación madre-feto durante el embarazo.
¿Qué procesos genéticos, fisiológicos y hormonales controlan el inicio del parto en humanos? En otros mamíferos, los niveles de progesterona son un factor esencial en el comienzo de las contracciones y el inicio del parto. Sin embargo, en humanos no es así. Los mecanismos que propician el inicio del parto en humanos todavía son algo desconocido. “Es un aspecto básico de la biología humana del que simplemente no sabemos la respuesta, y es un poco vergonzoso no saberla”, explica el Dr. Vincent Lynch, autor del estudio y biólogo evolutivo en la Universidad de Búfalo.
Los investigadores han identificado al gen HAND2 como gen implicado en el inicio del parto en humanos. Imagen: Ernesto del Aquila III (NHGRI, CC BY NC 2.0, https://creativecommons.org/licenses/by-nc/2.0/).
HAND2, un factor de transcripción activo en el endometrio de los mamíferos placentarios
Con el objetivo de determinar algunos factores genéticos implicados en el inicio del parto en humanos, los autores realizaron un análisis comparativo en el que se utilizaron datos del transcriptoma de diferentes especies animales. Estos datos, obtenidos de diferentes bases científicas, incluyen datos de mamíferos placentarios (como es el caso del ser humano), así como de mamíferos monotremas, marsupiales, aves, reptiles y anfibios.
Tras el análisis, los investigadores encontraron diferentes genes que, en mamíferos placentarios, evolucionaron para mantenerse activos durante el embarazo. Estos genes no modifican su actividad durante la reproducción en el resto de las especies estudiadas. Entre ellos, los autores destacan el gen HAND2.
HAND2, ubicado en el cromosoma 4 en humanos, codifica para un factor de transcripción relacionado con la supresión de la señalización del estrógeno. Se trata, además, de un gen pleiotrópico, que también se ha asociado al desarrollo del sistema cardiovascular. “HAND2, que ancestralmente juega un papel en el desarrollo del corazón, evolucionó para activarse en el útero durante el embarazo, cuando aspectos como la comunicación materno-fetal, la tolerancia inmune materna del feto antigénicamente distinto y la gestación interna prolongada son importantes.”, explica el Dr. Lynch. “Nuestro estudio sugiere que HAND2 podría estar involucrado en la facilitación de algunos de estos procesos”, añade.
HAND2 controla la expresión de la citocina IL-15 en fibroblastos del estroma endometrial
Para determinar el tipo de células que expresan HAND2 en el útero materno humano, los autores analizaron datos de la expresión de células individuales en deciduas en el primer trimestre de gestación. Gracias a ello, los autores determinaron que la expresión de HAND2 en humanos durante el embarazo se produce casi exclusivamente en los fibroblastos del estroma endometrial.
El siguiente paso de los investigadores fue analizar los cambios en la expresión de diferentes genes en fibroblastos del estroma endometrial carentes de la actividad de HAND2. El equipo encontró diferentes genes cuya actividad se vio alterada, entre ellos el gen IL15. Este gen codifica para una citoquina, la interleucina 15, una molécula capaz de inducir la proliferación de ciertas células del sistema inmunitario.
HAND2 durante el ciclo menstrual y el embarazo
En el estudio, los investigadores analizaron también los niveles de actividad de HAND2 durante el ciclo menstrual y la gestación en humanos. Los resultados demostraron que la actividad de HAND2 en el útero se ve aumentada a partir de la fase proliferativa del ciclo menstrual y alcanza un pico máximo a finales de la fase secretora. En cuanto a la gestación, los autores observaron que la actividad del gen se ve reducida a partir del tercer trimestre de embarazo, alcanzando niveles mínimos poco antes del comienzo del parto.
Los resultados de este nuevo estudio suponen una mejora en la comprensión de los factores genéticos que influyen en el desarrollo del embarazo y en el inicio del parto, si bien, todavía es necesario continuar investigando los factores que determinan la duración de la gestación. “Dada la posibilidad de que la expresión dinámica de HAND2 durante el transcurso del embarazo pueda ser importante en la regulación de la duración de la gestación, será emocionante seguir construyendo sobre nuestros resultados, especialmente en el contexto de la investigación del parto prematuro”, explica la Dra. Mirna Marinić, autora del estudio e investigadora en el Departamento de Genética Humana en la Universidad de Chicago.
Artículo original: Marinić M, et al. Evolutionary transcriptomics implicates HAND2 in the origins of implantation and regulation of gestation length. Elife. 2021 Feb 1;10:e61257. doi: 10.7554/eLife.61257. https://elifesciences.org/articles/61257
Las mutaciones producidas en las proteínas histonas que se relacionan con el cáncer aumentan la remodelación del material hereditario y pueden influir en los destinos celulares, concluye una reciente investigación de la Universidad de Princeton. Los resultados del trabajo, publicado en Nature Chemical Biology, proporcionan las nuevas claves de la participación de las mutaciones en las proteínas histonas en el cáncer y ofrecen una estrategia para responder a futuras cuestiones biológicas.
La organización del material hereditario en el interior del núcleo celular tiene un papel relevante en la replicación del ADN, la reparación de posibles del genoma y la actividad de los genes. En todos estos procesos es necesario que la maquinaria proteica responsable tenga acceso directo a la molécula de ADN, por lo que factores que influyan sobre cómo está compactado el ADN o cómo de accesible sea, pueden afectar tanto al mantenimiento del ADN como a la expresión génica.
Ciertas mutaciones en las proteínas histonas están asociadas a diferentes tipos de cáncer, así como a diferentes grados de la enfermedad. Algunas de estas mutaciones, denominadas mutaciones de oncohistonas, se localizan cerca de regiones de las proteínas histonas que sufren modificaciones tras la síntesis de la proteína y pueden influir en su función a través del conocido código epigenético de las histonas. Otras mutaciones se encuentran en regiones diferentes y los mecanismos por los que pueden influir en la función de las histonas se desconocen.
Las proteínas histonas tienen un papel estructural en la compactación del ADN e intervienen en diferentes procesos de mantenimiento y regulación de la expresión del genoma. Imagen: unidades de histonas que intervienen en la formación de los nucleosomas. David O Morgan, via Wikimedia Commons.
Un reciente estudio dirigido por los equipos de Tom Muir de la Universidad de Princeton y Davis Allis de La Universidad Rockefeller ha abordado la caracterización bioquímica y los efectos biológicos de aproximadamente 150 mutaciones en histonas identificadas en cáncer y seleccionadas por su frecuencia, evidencias previas y carga mutacional del tumor donde se encontraron.
Los investigadores han desarrollado dos herramientas de rastreo. En primer lugar, han utilizado una biblioteca de nucleosomas con mutaciones del cáncer marcados con etiquetas de ADN. Esta estrategia permite utilizar la secuenciación masiva para identificar los nucleosomas y evaluar a gran escala y a nivel bioquímico los efectos de las mutaciones en las histonas. A través de esta aproximación, el equipo pudo estimar cómo afecta cada mutación a la conformación del nucleosoma. En segundo lugar, los investigadores utilizaron una biblioteca de células de levadura que habían sido humanizada a través de la inserción de los genes humanos de las histonas, que permitió evaluar el impacto de las diferentes mutaciones en un contexto celular.
El análisis de las mutaciones en las histonas señala a tres zonas de la estructura del nucleosoma donde los efectos de las mutaciones son más pronunciados: aquellas donde interacciona con el ADN, una región denominada zona acidófila y otra porción estructural que afecta a su estabilidad.
Las mutaciones en las proteínas histonas que se relacionan con el cáncer aumentan la remodelación del material hereditario
Los resultados del trabajo presentan a las mutaciones de las histonas que ocurren en el dominio globular como potenciales participantes en el desarrollo del cáncer, a través de su remodelación de la cromatina y sus efectos sobre la expresión génica. Hasta el momento este tipo de mutaciones de las histonas no había recibido mucha atención.
“Encontramos mutaciones dentro del núcleo de las histonas, las cuatro histonas, y parecen hacer cosas completamente diferentes a las que hacen las mutaciones en histonas previamente identificadas”, señala John Bagert, investigador postdoctoral en el laboratorio de Muir y uno de los primeros autores del trabajo. “Afectan a la estructura real del nucleosoma y parecen desestabilizarlo en algunos casos”. Bagert señala que, hasta el momento, estas características de los nucleosomas no se habían relacionado con el cáncer. “Creemos que las mutaciones que afectan al remodelado de la cromatina pueden contribuir a la enfermedad y al cáncer en humanos”, señala el investigador. “Hemos identificados los sitios y mutaciones en esos sitios que pensamos que causan problemas”.
Los investigadores proponen un modelo de cómo los nucleosomas que pierden parte de su estabilidad como consecuencia de mutaciones en las histonas pueden promover el desarrollo del cáncer o su progresión. Las mutaciones afectan la expresión génica debido a la diferente accesibilidad de la maquinaria proteica responsable al ADN.
Una de las cuestiones que permanecen por resolver es en qué momento del desarrollo del cáncer se producen las mutaciones en las histonas relacionadas con esta enfermedad. Algunas de las mutaciones en histonas relacionadas con el cáncer se producen de forma temprana, pero se desconoce en qué estadio concreto. “No lo sabemos todavía”, señala Michelle Mitchener, investigadora postdoctoral en el laboratorio de Muir y una de los primeros autores del trabajo. “Una de las cosas más emocionantes es que hemos comenzado el proceso de descubrir lo que estas mutaciones hacen. Hemos identificado algunas nuevas posiciones que creo que deberían ser investigadas más. Hay todavía muchas cuestiones sobre los mecanismos”. Otras cuestiones pendientes de investigar son la relación de las mutaciones en las histonas con otras mutaciones relacionadas con el cáncer, el papel de las alteraciones de las histonas en otras enfermedades.
Por último, más allá de los resultados obtenidos en el estudio, las herramientas de análisis utilizadas en el estudio representan una plataforma experimental con gran potencial para investigar el efecto de mutaciones en histonas a gran escala. En este caso, han permitido evaluar el efecto de las mutaciones asociadas al cáncer en las histonas de forma detallada. Estas mismas aproximaciones podrían contribuir a responder nuevas preguntas sobre el papel de las mutaciones en las histonas en el cáncer. “La velocidad lo es todo”, señala Bagert. “Hay tanta biología que no podemos hacerlo todo. Tan solo necesitamos encontrar formas de hacerlo rápido. Creo que este es el futuro de la biología”.
Un estudio dirigido por investigadores de la Universidad de Barcelona, el Centro de Regulación Genómica y Instituto de Investigaciones Biomédicas August Pi y Sunyer demuestra el papel patogénico del ARN en el desarrollo de la enfermedad de Huntington y ofrece una nueva vía para investigar tratamientos para la enfermedad.
La enfermedad de Huntington es un trastorno neurodegenerativo hereditario causado por una expansión de trinucleótidos CAG en el gen HTT que codifica para la proteína huntingtina. Imagen cortesía de Dr. La Padula.
La enfermedad de Huntington es un trastorno neurodegenerativo hereditario causado por una expansión de trinucleótidos CAG en el gen HTT que codifica para la proteína huntingtina. En presencia de esta expansión, la proteína huntingtina resultante es más larga de lo normal, lo que tiene varias consecuencias: en primer lugar, se induce su degradación en fragmentos más pequeños que resultan tóxicos para la célula y, en segundo lugar, no se produce suficiente proteína HTT normal. A nivel clínico, los pacientes con enfermedad de Huntington presentan alteraciones motoras, cognitivas y psiquiátricas.
En paralelo a los estudios sobre la proteína huntingtina mutada, recientemente, se han descrito otros mecanismos, basados en ARN, que también intervienen en la patología de la enfermedad. Por ejemplo, la generación de fragmentos repetitivos de ARN derivados de la expansión de trinucleótidos CAG puede resultar perjudicial para las células. Además, se han detectado cambios en los niveles de otros ARNs de pequeño tamaño, aunque hasta el momento su papel en la enfermedad era incierto.
Para investigar cómo influyen esos otros ARNs pequeños en la patología del Huntington los investigadores aislaron una muestra de ellos a partir de cerebros de pacientes con la enfermedad y personas no afectadas y los inyectaron en el cerebro de ratones sanos. Al analizar estos animales el equipo encontró que los ARNs producidos en pacientes son capaces y suficientes para inducir la enfermedad. Los ratones que recibieron ARNs pequeños derivados de los pacientes mostraron problemas motores y cambios de expresión génica y proteínas compatibles con la patología de la enfermedad presente en humanos.
Los investigadores encontraron que el efecto neurotóxico de los ARNs pequeños podía ser mitigado hasta cierto punto a través del bloqueo específico de la expansión patológica de CAGs del gen HTT. Esto sugería que más allá de los ARNs repetitivos y pequeños generados de las repeticiones CAG debía haber otros tipos de ARN pequeño con potencial neurotóxico.
Con el objetivo de identificar a esos otros ARNs pequeños neurotóxicos los investigadores analizaron qué tipos de ARNs pequeños eran más frecuentes en el cerebro de los pacientes con enfermedad de Huntington y encontraron que los niveles de ARN derivados de ARNs de transferencia son los más alterados en la enfermedad. Además, también demostraron el potencial neurotóxico de uno de ellos en cultivos neuronales.
Los resultados del trabajo reafirman el papel de los ARNs pequeños no derivados del gen HTT en la patología de la enfermedad de Huntington y comienzan a ofrecer claves sobre su participación en la misma. “Los efectos tóxicos relativos a los ARNs con repeticiones CAG no explican ciertas alteraciones que son importantes en el contexto de la patología, como por ejemplo la afectación neuronal específica o las alteraciones transcripcionales”, señalan los autores del trabajo. “Estos resultados, muestran que diferentes tipos de ARNs pequeños producidos en los cerebros de los pacientes probablemente tienen un papel en la patogénesis”.
La presencia de estos ARNs pequeños, producidos alterados y funcionalmente importantes en la enfermedad de Huntington tienen relevancia para el futuro desarrollo de estrategias terapéuticas y para la identificación de nuevos biomarcadores de la enfermedad. En el primer caso, determinar los ARNs pequeños implicados abre una vía hacia la posibilidad de bloquear su efecto patológico. Y en el caso de los biomarcadores, la identificación de una alteración en los niveles de los ARNs pequeños podría favorecer el diagnóstico y tratamiento temprano de la enfermedad.
Por último, estos resultados no solo son importantes para la enfermedad de Huntington, sino que también podrían ser extrapolables y tener importantes implicaciones en el estudio de otras enfermedades neurodegenerativas.
Referencia: Creus-Muncunill J, et al. Huntington’s disease brain-derived small RNAs recapitulate associated neuropathology in mice. Acta Neuropathologica. 2021. DOI: https://doi.org/10.1007/s00401-021-02272-9
PUBLICADO EL MARZO 24, 2021 Rubén Megía González, Genotipia
Un reciente estudio, publicado el pasado 10 de marzo en Science Advances, ha identificado 50 nuevas regiones del genoma que están involucradas en el color de los ojos.
La coloración del iris está determinada por la abundancia de melanina en su epitelio pigmentario y la densidad y distribución de los melanocitos, factores a los que hay que añadir la proporción de los dos tipos de melanina (eumelanina y feomelanina) y la absorción y dispersión de la luz causada por componentes extracelulares. Imagen: v2osk vía Unsplash.
El color de ojos es uno de los rasgos físicos más característicos de una persona. La coloración de nuestro iris viene determinada por la abundancia de melanina en su epitelio pigmentario y la densidad y distribución de los melanocitos. Estos factores, sumados a la proporción de los dos tipos de melanina (eumelanina y feomelanina) y a la absorción y dispersión de la luz causada por componentes extracelulares, son los que diferencian cada una de las tonalidades descritas para el ojo humano. Anteriores estudios ya habían encontrado genes relacionados con el color de los ojos, como es el caso de HERC2 y OCA2.
Ahora, un equipo liderado por investigadores del King’s College de Londres y el Centro Médico de la Erasmus University Rotterdam, ha identificado 50 nuevos genes involucrados en el color del iris en humanos. El estudio, realizado en casi 195.000 personas, es el más amplio hasta la fecha.
En el estudio, los investigadores utilizaron datos del genoma completo de 157.485 individuos con ascendencia europea, obtenidos de las bases de datos de la empresa 23andMe. A este número total, se le sumaron los datos de otros 35.501 individuos de ascendencia europea y 1.636 individuos con ascendencia asiática en la fase de replicación del trabajo.
Tras el análisis del genoma completo de los casi 195.000 individuos, los autores encontraron un total de 50 loci relacionados con el color de ojos. Ocho de estos nuevos loci ya habían sido relacionados con otros rasgos de la pigmentación, como el color del pelo o el color de piel.
Los resultados de este estudio suponen una mejora en la comprensión de los factores genéticos implicados en algunos aspectos de la pigmentación del ojo. “Los descubrimientos son emocionantes porque nos acercan un paso más a comprender los genes que causan uno de los rasgos más impactantes del rostro humano, que ha desconcertado a generaciones a lo largo de nuestra historia”, explica el Dr. Pirro Hysi, autor del estudio e investigador en el Departamento de Oftalmología y en el Departamento de Estudios de Gemelos y Epidemiología Genética del King’s College de Londres. “Esto mejorará nuestro conocimiento sobre algunas enfermedades que sabemos que están asociadas con niveles específicos de pigmentación”, añade.
Otra de las aplicaciones de este estudio es en el ámbito de la genética forense. “Este estudio proporciona el conocimiento genético que se necesita para mejorar la predicción del color de ojos que actualmente se aplica en estudios antropológicos y forenses”, explica el Dr. Manfred Kayser, autor e investigador del Departamento de Identificación Genética del Centro Médico de la Erasmus University Rotterdam.
Artículo original: Simcoe M, et al. Genome-wide association study in almost 195,000 individuals identifies 50 previously unidentified genetic loci for eye color. Sci Adv. 2021 Mar 10;7(11):eabd1239. doi: http://dx.doi.org/10.1126/sciadv.abd1239.
Dos estudios publicados en Nature proporcionan un nuevo modelo para estudiar el desarrollo humano temprano. A partir de células madre pluripotentes o de la reprogramación de fibroblastos los investigadores han conseguido inducir la reorganización de las células en estructuras similares a los blastocistos humanos que, sin ser embriones reales, reproducen algunas de las características de las primeras etapas embrionarias.
Una de las primeras etapas del desarrollo es la formación del blastocisto, una estructura esférica que consta de diferentes tipos celulares que darán lugar al embrión, la placenta y otros tejidos. Si bien los diferentes pasos de este proceso se conocen a grandes rasgos, la investigación en detalle de lo que ocurre en las diferentes estructuras embrionarias humanas y sus efectos en estadios posteriores ha estado limitada en gran medida por la disponibilidad de modelos de estudio adecuados. Hasta el momento, dependía de la donación de blastocistos humanos obtenidos tras la fecundación in vitro, estrategia con diferentes retos técnicos y éticos.
Recientemente, se habían conseguido obtener estructuras similares a los blastocistos, denominadas blastoides, a partir de células madre de ratón, que han favorecido el estudio del desarrollo temprano en esta especie.
Ahora, dos estudios publicados en Nature presentan la obtención de blastoides humanos como una herramienta para mejorar el conocimiento sobre los primeros pasos del desarrollo humano. Dos equipos dirigidos por investigadores de la Universidad Monash de Australia y la Universidad de Texas Southwestern han conseguido mejorar aproximaciones las técnicas y de cultivo disponibles hasta el momento y generar blastoides humanos a partir de células madre humanas de diferente origen con los que es posible estudiar lo que ocurre en los primeros días tras la fecundación.
Imágenes de diferentes tinciones de los blastoides. Imagen: Monash University.
Origen celular diferente, pero características similares en los blastoides humanos
El equipo de la Universidad Monash ha obtenido blastoides a partir de células madre embrionarias humanas y células madre pluripotentes, derivadas de células adultas. Por otra parte, el equipo de la Universidad de Texas Southwestern ha generado blastoides a partir de células madre reprogramadas a partir de fibroblastos de la piel.
En ambos casos, los blastoides recapitulan características propias de los blastocistos humanos. Presentaban un tamaño, forma y número de células similar y se organizan según una estructura esférica, cavidad y formación de masa celular interna características. Además, los datos de expresión génica confirman la identidad de los diferentes tipos celulares.
Utilidad de los blastoides en la investigación
Los blastoides ya han empezado a proporcionar información relevante sobre el desarrollo humano. Dirigidos por Gary C Hon y Jun Wu, los investigadores de la Universidad de Texas Southwestern han encontrado que algunos miembros de la familia de la proteína kinasa C, que interviene en el ensamblaje y mantenimiento de ciertas uniones entre las células, regulan la formación de la cavidad de los blastoides humanos.
“Estudiar el desarrollo humano es muy difícil, especialmente en estos estadios”, ha señalado Wu en la rueda de prensa de presentación de los trabajos. “La información que hemos aprendido está basada en la utilización de embriones humanos, algo a lo que solo pueden acceder algunos laboratorios privilegiados. Nuestro modelo puede abrir un camino en este campo, que permita que cualquier laboratorio pueda generar un modelo donde probar sus hipótesis sin utilizar embriones humanos”. El investigador también destaca que, puesto que los blastoides se pueden producir en gran cantidad, se podrá realizar rastreos con múltiples agentes químicos o toxinas que puedan afectar al desarrollo humano durante las primeras etapas. Igualmente, se podrían realizar análisis genéticos para ver qué genes están interviniendo. Y, además, los blastoides podrían proporcionar una vía para estudiar in vitro la implantación del embrión, proceso para el que en estos momentos no se dispone de estrategia de investigación.
Un modelo adecuado, pero con limitaciones
Investigadores de la Universidad de Monash que han intervenido en uno de los estudios:Jia Tan, Jose Polo, Xiaodong (Ethan) Liu. Imagen: Monash University.
El desarrollo de blastoides ofrece un entorno tridimensional en el que estudiar el desarrollo temprano humano muy similar al de los embriones reales. Sin embargo, también ofrece algunas limitaciones como por ejemplo el hecho de que el proceso de obtención es largo y los blastoides no se producen de forma sincronizada o que hay variaciones en la eficiencia de producción según la línea celular o condiciones iniciales. “El modelo representado por los blastoides no es perfecto”, destaca Jun Wu. “Hay mucho que no sabemos sobre el desarrollo humano temprano. El modelo de blastoides nos proporciona parte de esa información, pero no toda. Como ocurre en el caso de ratones, los blastoides no son equivalentes a blastocistos”.
No son embriones humanos, pero ¿pueden plantear cuestiones éticas?
Los blastoides no son embriones. Los investigadores responsables de ambos estudios se mantienen firmes en esa consideración sobre el estatus de los blastoides humanos. Amander Clark, investigadora de la Universidad de California Los Angeles, quien ha participado en uno de los estudios, ha destacado que los blastoides son estructuras de células humanas similares a embriones, que ciertamente se parecen a los blastocistos y muestran algunas de sus características, pero que no pueden considerarse blastocistos. De hecho, siguiendo la normativa actual, deben dejar de cultivarse antes de que se forme la línea primitiva, que marca el inicio de la gastrulación. “Mucho antes de que se forme el sistema nervioso”, señala Clark.
Jun Wu los define como “una colección de células que experimentan los primeros pasos de la embriogénesis”. “Son un buen modelo que puede contribuir a sobrepasar algunos de los problema de la investigación actual”, indica el investigador.
De forma similar José Polo, investigador en el Departamento de Anatomía y Biología del Desarrollo de la Universidad Monash de Australia y director de uno de los estudios, señala que “las diferencias moleculares y el potencial funcional de los blastoides nos indican que no deberían ser considerados equivalentes a blastocistos, aunque sí son un sistema muy bueno para modelar algunos aspectos de la biología de los blastocistos”.
Dado que los dos tipos de blastoides humanos presentados en los estudios pueden ser obtenidos a partir de células somáticas adultas y no requieren la destrucción de embriones humanos reales, los investigadores anticipan que su uso tendrá una mayor aceptación como modelo de estudio. Además, los investigadores no contemplan utilizar esta estrategia para generar embriones que puedan ser implantados.
No obstante, tal y como destacan Yi Zheng y Jianping Fu, del Departamento de Ingeniería Mecánica de la Universidad de Michigan en un comentario que acompaña la publicación de los artículos, el desarrollo de los blastocistos se podría considerar un paso hacia la ingeniería de embriones humanos. Zheng y Fu señalan que el continuo desarrollo de modelos de estudio de embriones humanos requiere de una conversación pública sobre la relevancia científica de este tipo de investigaciones y los aspectos sociales y éticos que puede plantear.
Investigadores de la Universidad Hebrea de Jerusalén han demostrado que el análisis de ADN nucleosómico libre en sangre permite obtener información sobre procesos patológicos activos en tejidos afectados por el cáncer u otras enfermedades.
Cuando las células mueren, fragmentos de su material genético son liberados en la circulación. En los últimos años, diferentes aproximaciones han abordado el análisis de estos fragmentos como fuente de información sobre la salud del organismo para desarrollar aplicaciones en la práctica clínica. Como resultado, existen métodos para detectar ADN tumoral o ADN fetal en la sangre de un paciente o una madre embarazada, respectivamente, y es posible también identificar el origen de un tumor a partir de las marcas epigenéticas propias del tejido del que deriva el ADN tumoral.
En un trabajo publicado el pasado enero en Nature Biotecnology, el equipo dirigido por Nir Friedman y Ronen Sadeh de la Universidad Hebrea de Jerusalén, ofrece una nueva aproximación para obtener información a partir de ADN libre en sangre. En este caso, los investigadores han analizado nucleosomas, las unidades básicas de la cromatina, consistentes en complejos de ocho histonas envueltas por alrededor de 150 pares de bases del ADN.
La combinación de modificaciones en las histonas en un momento concreto influye en la accesibilidad de la maquinaria de expresión génica al ADN y marca la actividad del genoma. Teniendo en cuenta esta característica, los investigadores utilizaron, en muestras de sangre, una técnica conocida como inmunoprecipitación de la cromatina, que permite seleccionar nucleosomas con modificaciones de las histonas concretos (que reflejan estadios de actividad del genoma específicos). A continuación, el equipo analizó el ADN de los nucleosomas seleccionados como capturas moleculares de la actividad génica. En total, el equipo analizó alrededor de 250 muestras, correspondientes a 61 donantes sanos, cuatro pacientes con infarto de miocardio, 29 pacientes con enfermedades autoinmunes o metabólicas y 56 pacientes con carcinoma colorrectal metastásico.
Los investigadores han utilizado una biopsia líquida basada en el análisis de nucleosomas, con la que se obtiene diferente información sobre de qué tejido derivan y qué genes estaban activos. Imagen: Darryl Leja, National Human Genome Research institute (www.genome.gov)
Tras analizar el ADN nucleosómico, los investigadores pueden obtener información sobre la naturaleza de la enfermedad, como qué regiones estaban activas o inactivas según las modificaciones de las histonas, dónde se localiza o cómo de avanzada puede estar la enfermedad. En el estudio, el equipo detectó diferencias entre las muestras de controles sanos y pacientes. Por ejemplo, obtuvieron información sobre la diferente actividad transcripcional de las personas con enfermedades hepáticas y diferenciaron firmas moleculares específicas del cáncer.
La clave del análisis del ADN nucleosómico se encuentra en la información epigenética que contiene. Friedman resalta que, una vez que comprendieron que esta información puede permanecer en el ADN presente en la sangre, pensaron que podrían utilizar esos datos para determinar el origen del tejido o células muertas y los genes que estaban activos en esas células. “Basándonos en esos resultados podemos revelar detalles clave sobre la salud del paciente”, explica el investigador. “Somos capaces de entender mejor porqué las células murieron, si hay una infección o cáncer y en función de ello estar en mejor posición para determinar cómo está desarrollándose la enfermedad”.
Los autores del trabajo destacan que una característica única de la secuenciación de ADN nucleosómico seleccionado por ChIP es que el paso en el que se seleccionan los nucleosomas según las marcas de las histonas genera una representación biológicamente relevante del genoma y permite hacer preguntas concretas. Esta aproximación ofrece información que la secuenciación o análisis de metilación del ADN libre en sangre no puede proporcionar, como es ofrecer una imagen de la actividad transcripcional.
“Esperamos que esta aproximación nos permita diagnósticos más tempranos de enfermedades y ayude a los médicos a tratar pacientes de forma más efectiva”, señala Ronen Sadeh, uno de los directores del trabajo.
Referencia: Sadeh R, et al. ChIP-seq of plasma cell-free nucleosomes identifies gene expression programs of the cells of origin. Nat Biotech. DOI: https://doi.org/10.1038/s41587-020-00775-6
Un estudio del CSIC demuestra en animales que las nanopartículas cargadas con el péptido GSE4 resultan altamente efectivas para prevenir y revertir la enfermedad
Investigadores del Consejo Superior de Investigaciones Científicas (CSIC) han desarrollado una nueva y prometedora terapia que consigue prevenir y revertir la fibrosis pulmonar. La terapia consiste en nanopartículas cargadas en su interior con una molécula, el péptido GSE4, que disminuye el daño del ADN en las células afectadas con esta enfermedad. Según ha demostrado el estudio, publicado en la revista The FASEB Journal, el tratamiento ha probado su eficacia en animales y sería el primer paso para estudiar su efecto protector en pacientes de fibrosis pulmonar idiopática (FPI) y de otras enfermedades respiratorias irreversibles con alteración de los telómeros.
La fibrosis pulmonar idiopática es una enfermedad fibrótica letal asociada al envejecimiento, con una supervivencia media de 2 a 5 años. Suele aparecer hacia los 60 años de edad, pero existen formas familiares (genéticas) en edades más tempranas. La fibrosis aparece como una respuesta anormal de cicatrización tras un daño en los alveolos de los pulmones y se caracteriza por una sustitución progresiva del tejido sano por tejido cicatricial, lo que provoca una pérdida de la función pulmonar. Los mecanismos responsables de la reparación y regeneración defectuosas no se conocen bien, pero estudios recientes sugieren un posible papel del envejecimiento acelerado y el acortamiento de los telómeros en la aparición de la enfermedad, principalmente en las formas familiares.
“Los telómeros son pequeñas estructuras ubicadas al final de los cromosomas que protegen las secuencias de ADN dentro de cada célula que lo resguardan y ayudan a mantener su integridad”, explica Rosario Perona, investigadora del IIB-CSIC-UAM y autora principal del estudio. “Cada vez que la célula se divide, los telómeros se acortan, hasta que llega un punto en que no pueden ejercer su función protectora y la célula no puede dividirse. Se considera, por tanto, que la longitud de los telómeros es un indicador de envejecimiento”, añade.
El estudio se ha centrado en el péptido GSE4, que forma parte de una proteína del complejo telomerasa, y que puede proteger las células de otras enfermedades con efectos en la actividad telomerasa, como la disqueratosis congénita y ataxia telangiectasia. La telomerasa es capaz de realargar los telómeros de las células enfermas y conseguir que estas se recuperen. Los investigadores han utilizado una nanopartícula para encapsular la molécula GSE4 y poder llevarla al interior de las células y al tiempo estabilizarla en el torrente circulatorio.
“Para probar la eficacia del péptido hemos usado como modelo las células alveolares de pulmón de rata tratadas con un agente que provoca fibrosis, la bleomicina. Cuando tratamos estas con nanopartículas que contienen GSE4, se produce una reversión del proceso inflamatorio y fibrótico producido por la bleomicina. Al aumentar la actividad de la telomerasa, disminuyen la fibrosis y la inflamación y aumenta la regeneración del tejido alveolar, lo que indica una eficacia terapéutica de las nanopartículas cargadas con GSE4 en este modelo de fibrosis pulmonar experimental”, detalla la investigadora del CSIC.
Los resultados del estudio prueban la necesidad de seguir con las investigaciones para mejorar la vía y los vehículos de administración del tratamiento con GSE4 mediante otras formas de encapsulación, de modo que en el futuro se logre un tratamiento curativo para pacientes con fibrosis pulmonar.
A la izquierda, células pulmonares tratadas con bleomicina con marcadores de fibrosis en rosa. A la derecha, la inoculación de nanopartículas con GSE4 produce una reducción significativa de la fibrosis. / Rosario Perona (IIB-CSIC-UAM)
El fármaco, logrado por un proyecto europeo con participación del CSIC, ha recibido la aprobación de ‘medicamento huérfano’, lo que avala su eficacia y permitirá su desarrollo comercial
Un equipo coordinado por investigadores del Consejo Superior de Investigaciones Científicas (CSIC) ha desarrollado un nanomedicamento eficaz para tratar la enfermedad rara de Fabry, que causa complicaciones cardiovasculares y cerebrovasculares, e insuficiencia renal, con episodios de dolor grave. Se estima que afecta a 2,6 de cada 10.000 personas en la Unión Europea. Este medicamento, resultado del proyecto europeo Smart4Fabry, ha sido designado como medicamento huérfano por la Comisión Europea, lo que avala su eficacia y permitirá su desarrollo comercial.
Los medicamentos huérfanos son aquellos fármacos destinados a tratar enfermedades poco frecuentes, que la industria se resiste a desarrollar por razones financieras. Con este nuevo producto farmacéutico, el CSIC cuenta con cuatro medicamentos designados como huérfanos. Es la primera vez que se refiere a un medicamento nanoformulado.
Necesidad de nuevos tratamientos para la enfermedad
La enfermedad de Fabry representa el trastorno de almacenamiento lisosómico (almacenamiento de sustancias en el proceso de ‘digestión’ en el interior de las células) más frecuente. Está causada por la ausencia o deficiencia de una enzima (denominada GLA) que provoca la acumulación lisosómica de algunas sustancias en las células de una amplia variedad de tejidos. Los tratamientos actuales consisten en la administración intravenosa de la enzima, pero presentan una eficacia limitada y una mala biodistribución.
El fármaco que se ha desarrollado es una nueva nanoformulación de la enzima GLA (nanoGLA) que mejora la eficacia en comparación con el tratamiento de referencia con GLA no nanoformulada. “El producto liposomal de tercera generación que hemos desarrollado en el proyecto ha demostrado, a nivel preclínico, una eficacia mejorada, frente a los tratamientos de remplazo enzimático autorizados. Esta nueva nanomedicina ha demostrado que la estrategia con nanoliposomas resulta altamente exitosa” explica Ibane Abasolo investigadora de CIBER-BBN y del VHIR, responsable de los estudios de eficacia en el proyecto.
El Comité de Medicamentos Huérfanos (COMP), de la Agencia Europea de Medicamentos (EMA), ha considerado que estos resultados constituyen una ventaja clínicamente relevante frente a los tratamientos de remplazo enzimático actuales. La designación de medicamento huérfano, además de ser un reconocimiento al beneficio significativo que ofrece la nueva nanomedicina frente a los productos ya autorizados para la enfermedad de Fabry, supone importantes implicaciones en la transferencia y en la traslación del nuevo producto terapéutico hacia fases más avanzadas de desarrollo.
Además, supone recibir una autorización de comercialización durante 10 años en los que no pueden comercializarse productos similares, y disponer de protocolos de asistencia y consejo científico gratuitos o con un coste reducido.
Investigadores de la universidad de UT Southwestern han encontrado que en pájaros cantores el gen Foxp1, relacionado previamente con el trastorno del espectro autista, interviene en la formación de memorias necesarias para ejecutar correctamente el canto. Los resultados del trabajo, publicado en Science Advances, muestran que la capacidad para formar memorias es un aspecto esencial para la transmisión cultural de comportamientos complejos como el lenguaje, que se adquieren por imitación y repetición. Además, plantean posibles mejoras en el desarrollo de terapias de comportamiento y comunicación para los niños con trastorno del espectro autista.
De forma similar a lo que ocurre en los humanos cuando aprenden a hablar, los pájaros cantores como el diamante mandarín aprenden o memorizan un conjunto de sonidos vocales y los empiezan a practicar imitando el canto de los adultos, más experimentados. Imagen: UT Southwestern Medical Center.
La imitación es un proceso esencial en el aprendizaje de ciertos comportamientos sociales y culturales, como por ejemplo el lenguaje. Los niños pequeños aprenden a reconocer e imitar los patrones de sonidos que utilizan los mayores cuando se comunican e incorporan esa información durante su adquisición del lenguaje.
En diversos trastornos del neurodesarrollo, como es el caso de los trastornos del espectro autista, la adquisición de algunos de estos comportamientos sociales está comprometida, aunque no siempre se conoce cuál es el mecanismo que está actuando.
Con el objetivo de profundizar más en esta cuestión el equipo de la Universidad de UT Southwestern se planteó si los genes relacionados con el autismo pueden afectar al comportamiento de imitación y formación de memorias que requiere la adquisición del lenguaje.
Concretamente, los investigadores decidieron investigar si el gen FOXP1 estaba implicado en este proceso. Este gen codifica para un factor de transcripción cuyas mutaciones aumentan el riesgo a desarrollar trastornos del espectro autista y, además, las personas que muestran una única copia funcional de FOXP1 desarrollan un síndrome característico que incluye diversos problemas con el lenguaje.
Pájaros cantores para estudiar la adquisición del lenguaje
Para investigar el papel de FOXP1 en los comportamientos repetitivos que ocurren durante la adquisición del lenguaje, el equipo recurrió a un modelo animal muy particular: los pájaros cantores, ya que, de forma similar a lo que ocurre en humanos cuando aprenden a hablar, los pájaros cantores como el diamante mandarín aprenden o memorizan un conjunto de sonidos vocales y los empiezan a practicar imitando el canto de los adultos, más experimentados.
Los investigadores encontraron que en el diamante mandarín, pájaros cantor utilizado como modelo, el gen FoxP1 se expresa en las neuronas del Centro Vocal Superior, una región cerebral conocida por contener neuronas espejo, un tipo de célula nerviosa importante para la imitación de acciones como la emisión de sonidos y el aprendizaje de melodías.
El equipo observó que, al inactivar el gen en esas neuronas, se bloquea la capacidad de los pájaros de aprender de los adultos. Además, la ausencia de FoxP1 afecta específicamente a la capacidad de los animales para formar memorias y no al comportamiento de imitación, otro componente importante del aprendizaje de nuevas melodías. Cuando la inactivación de Foxp1 se producía después de que los pájaros cantores hubieran creado memorias a partir de las melodías de sus tutores, el comportamiento de repetición de no se veía afectado y las canciones no se diferenciaban de los animales control.
A partir de estos resultados, los investigadores se plantearon que la actividad de FoxP1 en el Centro Vocal Superior no es necesaria para aspectos motores del aprendizaje del canto, sino que es específica e implica la formación de memorias.
Para confirmar sus resultados el equipo analizó la actividad y función de neuronas del Centro Vocal Superior, encontrando que la inactivación de FoxP1 afecta a la plasticidad de estas neuronas e induce otros cambios asociados a la formación de memorias.
Resultados relevantes para el conocimiento sobre los trastornos del espectro autista
Los resultados del trabajo ofrecen nueva información sobre cómo se transmiten los comportamientos repetitivos de una generación a la siguiente y cómo la alteración de los mecanismos implicados puede ocasionar trastornos concretos.
“Nuestros resultados sugieren que FoxP1 es clave en la formación de memorias de melodías en estos pájaros, que son críticas para la imitación posterior en su vida,” señala Todd Roberts, profesor de neurociencia en el Instituto del Cerebro Peter O´Donell Jr de la Universidad UT Southwestern y director del trabajo. “Un déficit similar en humanos podría jugar un papel paralelo en el desarrollo del lenguaje, bloqueando a los bebés en su capacidad para formar memorias del habla de los adultos que escuchan a su alrededor y comprometiendo su propia comunicación conforme crecen”.
Los autores del estudio indican que la información obtenida podría ser relevante para el desarrollo de nuevas terapias para los niños con trastorno del espectro autista. Algunas terapias actuales están centradas en ayudar a aprender las habilidades motoras necesarias para producir sonidos. Sin embargo, la formación de memorias podría ser más importante, como ocurre en el caso de los pájaros cantores. Esta consideración podría mejorar las terapias de comportamiento y comunicación para los niños con trastorno del espectro autista. Además, según señala Roberts, en el futuro podría llegar a ser plantearse la modificación del gen FoxP1 o la recuperación farmacológica de su función como potencial estrategia para evitar los déficits en el lenguaje. Estudios futuros deberán abordar estas cuestiones.
Por último, el trabajo aporta una nueva aproximación para estudiar el papel de los genes en el desarrollo de algunas de las características del trastorno del espectro autista. “Este estudio no solo es crítico para entender los síntomas de pacientes con trastorno del espectro autista asociado a FOXP1, sino que también sienta una base de trabajo para estudiar muchos otros genes asociados a los trastornos del espectro autista utilizando el sistema de pájaros cantores”, destaca Genevieve Konopka, profesora de neurociencias en la Universidad UT Southwestern y una de las autoras del trabajo.
Referencia: García-Oscos F, et al. Autism-linked gene FoxP1 selectively regulates the cultural transmission of learned vocalizations. Science Advances. 2021. DOI: http://dx.doi.org/10.1126/sciadv.abd2827
Investigadores del Centro Nacional de Investigaciones Oncológicas han encontrado que, independientemente de la edad, los pacientes con formas más graves de COVID-19 presentan telómeros más cortos que el resto de pacientes. El equipo plantea que el acortamiento de los telómeros es una consecuencia de la infección vírica e impide la regeneración de los tejidos.
Cromosomas con telómeros marcados. Imagen: Hesed Padilla-Nash y Thomas Ried (Instituto Nacional de Salud, EE.UU.).
Las estructuras terminales de los cromosomas, o telómeros, son esenciales para mantener la integridad del material hereditario en las células. Desde hace años, se conoce la relación entre su longitud y el envejecimiento, de forma que su acortamiento con las diferentes divisiones que sufren las células para mantener o regenerar los tejidos, es una marca distintiva del envejecimiento.
El Grupo de Telómeros y Telomerasa del CNIO, que lidera María Blasco lleva décadas investigando la función de los telómeros y su relación con diversas enfermedades. Una de sus áreas de investigación es la fibrosis pulmonar, enfermedad caracterizada por la progresiva formación de cicatrices en el tejido pulmonar y consecuente pérdida de función respiratoria. A través de diferentes estudios, el equipo de Blasco ha demostrado que la fibrosis pulmonar está causada por la presencia de daños en los telómeros de las células responsables de regenerar el tejido pulmonar, los neumocitos alveolares de tipo II.
La infección por coronavirus fuerza la capacidad regenerativa de los tejidos para hacer frente a los daños y compromete su equilibrio. Los neumocitos alveolares de tipo II se encuentran dentro de las diferentes células del organismo que ser infectadas por el virus, lo que unido a algunas de las manifestaciones clínicas de la enfermedad, como es la formación de fibrosis en los pulmones, llamó la atención del equipo de Blasco ““Cuando leí que en la COVID-19 estaban implicados los neumocitos alveolares tipo II, enseguida pensé que los telómeros podían tener un papel”, señala Blasco.
Los investigadores se plantearon si la infección por coronavirus podría estar agotando el potencial regenerativo de los tejidos y aquellas personas con telómeros cortos, y por lo tanto menos capacidad regenerativa, tendrían una mayor susceptibilidad a tener lesiones pulmonares y desarrollar formas más graves de la enfermedad.
“Sabemos que el virus infecta a los neumocitos alveolares tipo II y que esas son las células relevantes para regenerar el pulmón; también sabemos que si tienen daño telomérico no pueden regenerar, induciendo fibrosis”, indica Blasco. “Esto es lo que se ve en pacientes con lesiones pulmonares tras la COVID-19: pensamos que desarrollan fibrosis pulmonar porque tienen telómeros más cortos y esto limita la capacidad regenerativa de sus pulmones”.
Para comprobar si su hipótesis era cierta, los investigadores analizaron la longitud de los telómeros de 89 pacientes ingresados en el hospital de campaña del IFEMA, en Madrid. El equipo encontró que, como era de esperar, los pacientes de mayor edad presentaban telómeros más cortos, lo que derivaba en que la severidad de la enfermedad (que afecta más a los más mayores) también estuviera relacionada con la longitud más corta de los telómeros. Además, los investigadores encontraron que, independientemente de la edad, los telómeros de los enfermos más graves de COVID-19 eran más cortos que los del resto de pacientes.
Los resultados del trabajo, publicado en Aging indican que la presencia de telómeros cortos podría influir en la severidad de la acción del coronavirus en el organismo. El equipo planea demostrar esta relación en experimentos en ratón, donde compararán la aparición de fibrosis debida a la infección por el coronavirus en ratones con telómeros cortos y ratones con telómeros normales.
Si la relación entre los telómeros y la gravedad de la COVID-19 se confirma, podría representar una potencial vía terapéutica a través de la activación de la función telomerasa en el tejido afectado. Esta posibilidad se apoya en estudios previos del equipo en ratón que han demostrado el potencial de la activación de la enzima telomerasa para el tratamiento de la fibrosis pulmonar asociada al acortamiento de los telómeros. Precisamente en esta línea el pasado año el CNIO y la Universidad Autónoma de Barcelona crearon una empresa, que tiene como objetivo el desarrollo de terapias génicas dirigidas a la telomerasa que puedan utlizarse en patologías relacionadas con el acortamiento de los telómeros.
“Dado que los telómeros cortos pueden ser alargados de nuevo mediante la telomerasa y que en trabajos previos hemos mostrado que la activación de la telomerasa tiene efecto terapéutico para retrasar el envejecimiento y en enfermedades de la edad, así como en enfermedades relacionadas con telómeros cortos, como la fibrosis pulmonar, es tentador especular que esta terapia podría mejorar algunas de las patologías que quedan en pacientes de Covid-19 una vez superada la infección viral, como las patologías en los pulmones similares a la fibrosis” concluyen los autores en el trabajo.
Referencia : Sánchez-Vázquez R, et al. Shorter telomere lengths in patients with severe COVID-19 disease. Aging. 2020. DOI: https://doi.org/10.18632/aging.202463
Un reciente estudio, publicado en la revista Immunity el pasado mes de febrero, ha identificado variantes genéticas que influyen en la respuesta a la inmunoterapia en 22 regiones del genoma humano.
El sistema inmunitario es una compleja red de sustancias, células, tejidos y órganos que defiende el organismo frente a infecciones o anomalías celulares. De forma natural, este sistema es capaz de detectar la aparición de células cancerosas y eliminarlas, evitando así su proliferación. No obstante, en ocasiones las células cancerosas pueden pasar desapercibidas por el sistema inmunitario, provocando la aparición de tumores.
Linfocitos T killer atacando una célula tumoral. Imagen: Alex Ritter, Jennifer Lippincott Schwartz and Gillian Griffiths, National Institutes of Health.
El estudio, realizado por investigadores de diferentes universidades, ha analizado el genoma completo y el microambiente tumoral de alrededor de 9.000 pacientes oncológicos con 30 tipos de tumores diferentes. Gracias a ello, los autores han podido determinar diferentes factores que influyen en la respuesta inmunitaria.
Los datos de los casi 9.000 pacientes fueron obtenidos de las bases de datos del Proyecto Atlas del Genoma del Cáncer (TCGA, por sus siglas en inglés).
Heredabilidad de los parámetros inmunológicos
En primer lugar, los investigadores estudiaron la influencia genética sobre la variación (también conocida como heredabilidad) de 139 parámetros inmunológicos, relacionados con diferentes aspectos del sistema inmunitario. Los resultados indicaron una alta heredabilidad para las estimaciones de infiltración de células NK y células T, dos tipos de linfocitos que conforman el sistema inmunitario, en los tumores. Otros factores que presentaron una alta heredabilidad son los componentes de la vía de señalización de los interferones, unas proteínas señalizadoras que ayudan al sistema inmunitario a detectar infecciones.
Variantes genéticas que influyen en la respuesta a inmunoterapias
La inmunoterapia es un tipo de tratamiento frente al cáncer que se basa en la acción antitumoral propia del sistema inmunitario del paciente. Una de las principales limitaciones de este tipo de terapias es que no son efectivas en todos los pacientes, ya que algunos de ellos no responden correctamente al tratamiento. Esta respuesta puede estar condicionada por diferentes factores tanto ambientales como genéticos.
En el estudio, los investigadores examinaron cómo los parámetros inmunológicos eran influidos por casi 11 millones de variantes génicas en los casi 9.000 pacientes. El equipo identificó 22 regiones del genoma cuyas variantes influyen de forma significativa en la respuesta a la inmunoterapia. En estas regiones se encuentran los genes IFIH1 y STING1, entre otros.
El gen IFIH1, ubicado en el cromosoma 2 humano, codifica para la subunidad 1 de una proteína receptora del sistema inmune. Los autores observaron que las variaciones en este gen pueden influir de diferente manera en la respuesta a las inmunoterapias. Por ejemplo, los pacientes con una variante de IFIH1 que predispone a la aparición de diabetes tipo 2, presentaban unos tumores con mayor inflamación, que responderían mejor a la acción de las inmunoterapias. Otras variantes tienen el efecto contrario, por lo que influirían negativamente en la acción de estas terapias.
El gen STING1 se encuentra en el cromosoma 5 humano y codifica para una proteína de membrana relacionada con la respuesta inmune innata. Estudios anteriores ya habían descrito ciertas variantes de este gen que influyen positivamente en la respuesta a las inmunoterapias. Los autores de este nuevo estudio han descubierto que ciertas variantes pueden afectar negativamente a la respuesta a inmunoterapias, haciendo a sus portadores menos propensos a responder a ellas.
Factores asociados a la respuesta inmunitaria
Para conocer mejor el papel del genoma en el desarrollo de la respuesta inmunitaria, los investigadores realizaron un análisis de variantes raras, que se presentan en una frecuencia reducida, en los 9.000 pacientes de las bases de datos del TCGA.
Los autores encontraron variantes genéticas relacionadas con la respuesta inmunitaria en genes relacionados con 10 categorías funcionales diferentes, como la vía de señalización WNT, la estabilización de los telómeros o la oncogénesis. Es el caso de los genes BRCA1, típicamente asociado a un mayor riesgo de padecer cáncer, y RLB1, un gen supresor de tumores.
Los resultados de este estudio se suman a los de otros estudios, como el publicado el pasado 27 de enero en la revista Cell. En este último, un equipo de investigadores del Instituto Francis Crick, del Instituto de Cáncer de la University College de Londres y del Centro de Excelencia para la Investigación del Cáncer de Pulmón de Cancer Research UK, identificó ciertos cambios genéticos en los tumores que permiten predecir la efectividad del tratamiento con inmunoterapia en pacientes individuales.
Ambos estudios ayudan a mejorar el conocimiento sobre los factores genéticos que influyen en la respuesta a inmunoterapias, a la vez que aportan detalles sobre las variantes genéticas que influyen en la respuesta antitumoral del sistema inmunitario. “Hay algunos factores que ya se habían asociado previamente con cómo de bien se responde a los tumores”, explica el Dr. Elad Ziv, profesor de Medicina en la Universidad de California en San Francisco y autor del estudio. “Lo que ha sido menos estudiado es cómo tu genética predice la respuesta inmune de tu sistema frente al cáncer”, añade Ziv.
El siguiente paso, tal y como explica el Dr. Ziv, es utilizar estos datos para predecir qué pacientes oncológicos pueden beneficiarse de las inmunoterapias actuales y desarrollar nuevos fármacos para aquellos que no. “Aún está lejos”, añade, “pero es lo que esperamos que salga a partir de este trabajo”.
Artículo: Sayaman RW, et al. Germline genetic contribution to the immune landscape of cancer. Immunity. 2021 Feb 9;54(2):367-386.e8.
Sayaman RW, Saad M, Thorsson V, Hu D, Hendrickx W, Roelands J, Porta-Pardo E, Mokrab Y, Farshidfar F, Kirchhoff T, Sweis RF, Bathe OF, Heimann C, Campbell MJ, Stretch C, Huntsman S, Graff RE, Syed N, Radvanyi L, Shelley S, Wolf D, Marincola FM, Ceccarelli M, Galon J, Ziv E, Bedognetti D. Germline genetic contribution to the immune landscape of cancer. Immunity. 2021 Feb 9;54(2):367-386.e8. doi: 10.1016/j.immuni.2021.01.011. PMID: 33567262.
Fuente: Response to Cancer Immunotherapy May Be Affected by Genes We Carry from Birth. University of California San Francisco. https://www.ucsf.edu/news/2021/02/419786/response-cancer-immunotherapy-may-be-affected-genes-we-carry-birth
Investigadores de la Universidad del Estado de Pensilvania, muestran en un artículo recientemente publicado en Nucleics Acids Research que el ADN con una conformación diferente a la de doble hélice B muestra una mayor tasa de mutaciones. Los resultados colocan a este tipo de ADN como una fuente importante de variación de la tasa mutacional en el genoma, con implicaciones médicas y evolutivas.
Aproximadamente un 13{5f4e897c6ee23822ae50e890000b794346543cd330c4e8aa45664bf184d11e2e} del ADN se encuentra organizado o plegado en conformaciones diferentes a la doble hélice B, que se considera la estructura canónica o predominante del ADN. Dentro de estas conformaciones se encuentra la formación de estructuras cruciformes, el ADN-Z, el ADN-H o estructuras G4, entre otros.
Las regiones del genoma con conformaciones alternativas a la doble hélice B intervienen en diferentes funciones de la célula: regulan la expresión génica, actúan como sitios de unión a proteínas, participan en el mantenimiento de los telómeros y en el control de la metilación. Además, también se han relacionado con diferentes enfermedades, aunque los mecanismos por los que estas estructuras pueden influir en el desarrollo de un proceso patológico no se conocen en detalle.
Estudios previos del equipo de investigación de la Universidad del Estado de Pensilvania habían revelado que, en sistemas artificiales, la maquinaria de replicación del ADN comete un mayor número de errores cuando tiene que copiar ADN con conformaciones alternativas a la doble hélice B. Los investigadores pensaron que esta situación podía deberse a que estas estructuras dificultan la lectura de la secuencia de ADN y se plantearon determinar si ocurría lo mismo en las células.
Los resultados del trabajo indican que el ADN en diferentes conformaciones a la hélice B tiene una mayor tasa de mutación. Imagen: Wilfried Guiblet y Dani Zemba, Penn State.
Influencia de la estructura del ADN en la tasa de mutación
El equipo comparó la tasa de mutación de las estructuras de ADN en hélice B respecto al resto de conformaciones en dos contextos. Primero consideró la variabilidad dentro de la propia especie humana, la variación más reciente, a través del análisis de polimorfismos de un único nucleótido o regiones variables de mayor tamaño. En segundo lugar, como medida de divergencia entre especies comparó los genomas de referencia humano y de orangután.
Los resultados indican que la mayor parte de estructuras alternativas a la doble hélice B presentan una tasa de mutación aumentada. “Cuando miramos en todos los factores conocidos que influyen en la variación regional de las tasas de mutación a lo largo del genoma el ADN no B es el mayor contribuyente”, destaca Francesca Chiaromonte, profesora de Estadística para las Ciencias de la Vida en la Universidad del Estado de Pensilvania y codirectora del trabajo. “Hemos estado estudiando la variación regional en las tasas de mutación durante mucho tiempo desde diferentes ángulos. El hecho de que el ADN no B contribuye tanto en esta variación es un descubrimiento importante”.
Resultados con implicaciones a en el estudio de enfermedades y análisis evolutivos
Los investigadores indican que los datos obtenidos son relevantes tanto a nivel médico como evolutivo.
A nivel médico la diferencia de tasa de mutación entre las diferentes conformaciones del ADN proporciona una conexión física a la influencia de diferentes regiones del genoma con la aparición de ciertas enfermedades. En este sentido, los autores del trabajo señalan que “estudios futuros deberían considerar el potencial de un locus para formar ADN no B cuando evalúan variantes genéticas candidatas para enfermedades genéticas humanas”.
Por otra parte, a nivel evolutivo, los resultados presentan a las conformaciones de ADN alternativas a la hélice B y su tasa de mutación elevada como fuentes de variación genética, condición necesaria para la evolución y adaptación de las especies. “Habitualmente se considera que las mutaciones ocurren de forma tan poco frecuente que, cuando se ve la misma mutación en diferentes individuos, se asume que esos individuos compartieron un ancestro que pasó la mutación a ambos”, señala Kateryna Makova, profesora de la Universidad del Estado de Pensilvania y codirectora del trabajo. “Sin embargo, es posible que la tasa de mutación sea tan elevada en algunas de estas regiones de ADN B que la misma mutación pueda ocurrir de forma independiente en diferentes individuos. Si esto es cierto podría cambiar la forma en la que pensamos sobre evolución”.
Artículo científico: Guiblet WM, et al. Non-B DNA: a major contributor to small- and large-scale variation in nucleotide substitution frequencies across the genome.Nucleic Acids Research. 2021. DOI: https://doi.org/10.1093/nar/gkaa1269
Un equipo de investigadores e investigadoras liderado por Carmen Espinós del Centro de Investigación Príncipe Felipe (CIPF) y por Sergio Aguilera-Albesa, del Complejo Hospitalario de Navarra (CH Navarra), describen una nueva forma clínica de ataxia causada por mutaciones en la β-III espectrina, que se caracteriza por ser una ataxia congénita no progresiva y neurodegenerativa (NPCA, non-progressive cerebellar ataxia).
Resumen gráfico de los resultados del trabajo.
Las mutaciones se detectaron mediante un panel de genes diseñado en el Laboratorio de Enfermedades Raras Neurodegenerativas del CIPF. Con el propósito de demostrar que las mutaciones eran realmente patológicas, se realizaron estudios de expresión de la proteína con las mutaciones, observando que éstas afectaban seriamente a la estructura de la proteína.
Los pacientes se evaluaron clínicamente en la Unidad de Neuropediatría del CH Navarra. Se observó que presentaban hipotonía, retraso en el desarrollo, síndrome cerebeloso y déficits cognitivos. Se trata de una grave enfermedad rara pediátrica.
Estos resultados permiten dar un paso más en la comprensión de las causas moleculares de enfermedades raras como la ataxia, pero también de otras enfermedades. Estos descubrimientos contribuyen a una mejor comprensión del fenotipo asociado a SPTBN2 y las mutaciones y mecanismos subyacentes en esta enfermedad.
Los hallazgos logrados muestran que el gen codificante de la β-III espectrina causa este nuevo fenotipo clínico y, por tanto, se debe considerar su estudio en pacientes con NPCA. “Es un estudio traslacional que mejora el consejo genético de pacientes con enfermedades raras y posibilita una mejor planificación familiar”, señala la investigadora del CIPF, Carmen Espinós.
“Si consultamos la base de datos online sobre enfermedades hereditarias, el OMIM (Online Mendelian Inheritance in Men), conocemos poco más de 4.000 genes implicados en enfermedad humana con certeza y, por tanto, sólo tenemos clara la implicación de aproximadamente el 22{5f4e897c6ee23822ae50e890000b794346543cd330c4e8aa45664bf184d11e2e} de los genes que codifican para proteínas, considerando que el genoma humano tiene 20.000 genes. Queda mucho trabajo por hacer para entender las bases moleculares de las enfermedades hereditarias”, explica la investigadora valenciana.
“Trabajos como el nuestro aportan conocimiento a esta área de investigación traslacional”, concluye Espinós.
Sobre las ataxias congénitas
Las ataxias congénitas no progresivas conforman un grupo heterogéneo de procesos ligados a factores diversos con signos inespecíficos como hipotonía precoz, dificultades para la masticación o retraso en adquisiciones motrices, los signos de afectación cerebelosa se ponen de manifiesto con el desarrollo o pueden estar ausentes cuando la afectación es muy grave.
Los avances en investigación genética molecular permiten una mejor categorización, porque todavía la mayoría de los casos siguen siendo desconocidos tanto en su etiología como su carácter hereditario.
Referencia: Sancho P, et al. Expanding the β-III Spectrin-Associated Phenotypes toward Non-Progressive Congenital Ataxias with Neurodegeneration. Int J Mol Sciences. 2021. DOI: https://doi.org/10.3390/ijms22052505
La investigación que desarrolla Anna Adam en INCLIVA gracias a una ayuda concedida por la Asociación Española Contra el Cáncer en Valencia ha confirmado que la molécula “microRNA-30b” es un indicador diagnóstico de cáncer de mama, lo que facilitaría la detección de este tipo de cáncer en fases muy iniciales.
Este nuevo hallazgo, publicado en ESMO Open, también ha concluido que la presencia de mayores niveles de estas moléculas en plasma sanguíneo se asocia a un estadío avanzado (metástasis) de este tipo de cáncer.
Cada año más 33.300 mujeres se diagnostican de cáncer de mama, siendo el tumor más frecuente en las mujeres españolas. La COVID-19 ha supuesto un incremento de la vulnerabilidad económica en este perfil de pacientes. Continuar leyendo «Una llave para la detección precoz de cáncer de mama»
Pese a que los neandertales se extinguieron hace alrededor de 40.000 años, parte de su legado genético, una pequeña fracción de genoma neandertal, derivada del contacto reproductivo de los neandertales con los humanos modernos, sigue presente en el genoma de muchas personas de origen euroasiático. Esta herencia neandertal, que puede suponer entre un1,5 y un 4 por ciento del genoma, tiene un impacto en la biología de los humanos actuales: algunas de sus variantes se han relacionado con enfermedades de la piel, la tolerancia al dolor, un mayor riesgo a tener depresión… y también con la respuesta a la infección con el coronavirus SARS-CoV-2.
La imagen completa del papel de la herencia neandertal en la respuesta a COVID-19 no se conoce todavía. Los primeros datos apuntan a que la influencia neandertal en la infección causada por SARS-CoV-2 es compleja y no siempre en la misma dirección, lo que implica que algunas variantes de origen neandertal podrían influir negativamente sobre el curso de la enfermedad y mientras que otras tienen un efecto protector.
La herencia genética neandertal influye en la aparición de diferentes enfermedades. Ahora, diversos estudios analizan su impacto en la progresión de COVID-19.
Herencia genética neandertal asociada a COVID-19 más grave
Una nueva técnica permite secuenciar y fotografiar, de forma simultánea, el ADN de una célula intacta o incluso de un embrión completo.
Obtener la secuencia del ADN de las células es posible mediante el uso de técnicas genómicas. En la imagen, fibroblastos de ratón, con su esqueleto celular (rojo), mitocondrias (verde) y ADN (azul). [Flickr/NIH]
Un estudio, publicado en tiempo reciente por la revista Science, describe un método que combina microscopia y análisis genómico con el objeto de obtener la organización y secuencia del ADN dentro de células intactas.
Fei Chen y su equipo, del Instituto Broad del Instituto Tecnológico de Massachussets y la Universidad de Harvard, fijaron fibroblastos humanos a fin de preservar su estructura. A continuación, añadieron una enzima que cortó los genomas de las células e insertaron pequeñas porciones de ADN en los fragmentos resultantes, que permanecieron en su ubicación original dentro de las células. Las porciones de ADN insertadas ayudaron a hacer millones de copias de los fragmentos genómicos, así como a agregarles códigos de barras de ADN únicos. Estos son pequeñas regiones de un gen, elegidas por consenso, que se utilizan como herramienta de identificación, de modo similar a cómo los códigos de barras sirven para identificar los productos en el supermercado Continuar leyendo «Cómo hacer un mapa en tres dimensiones de un genoma»
Investigadores del Georgia Institute of Technology and Emory University han utilizado con éxito una estrategia basada en CRISPR para reducir la infección por el virus de la gripe y el coronavirus SARS-CoV-2 en modelos animales.
La estrategia antiviral diseñada por los investigadores consiste en hacer llegar a las células afectadas por la infección el ARN mensajero de una enzima que corta el ARN del genoma viral en posiciones específicas y conservadas. Tras el tratamiento los investigadores han observado una reducción en la concentración de virus de la gripe en ratones y una reducción en la replicación del coronavirus y síntomas de la enfermedad en hámsteres. La posibilidad de cambiar un componente del tratamiento para dirigirlo de forma específica a diferentes virus, ofrece la posibilidad de adaptar la estrategia a otros virus de forma rápida. Esta aproximación podría resultar de gran utilidad en el caso de virus emergentes, una vez obtenida la secuencia de su genoma. Continuar leyendo «Una estrategia basada en CRISPR muestra resultados prometedores frente al virus de la gripe y el coronavirus SARS-CoV-2 en modelos animales»
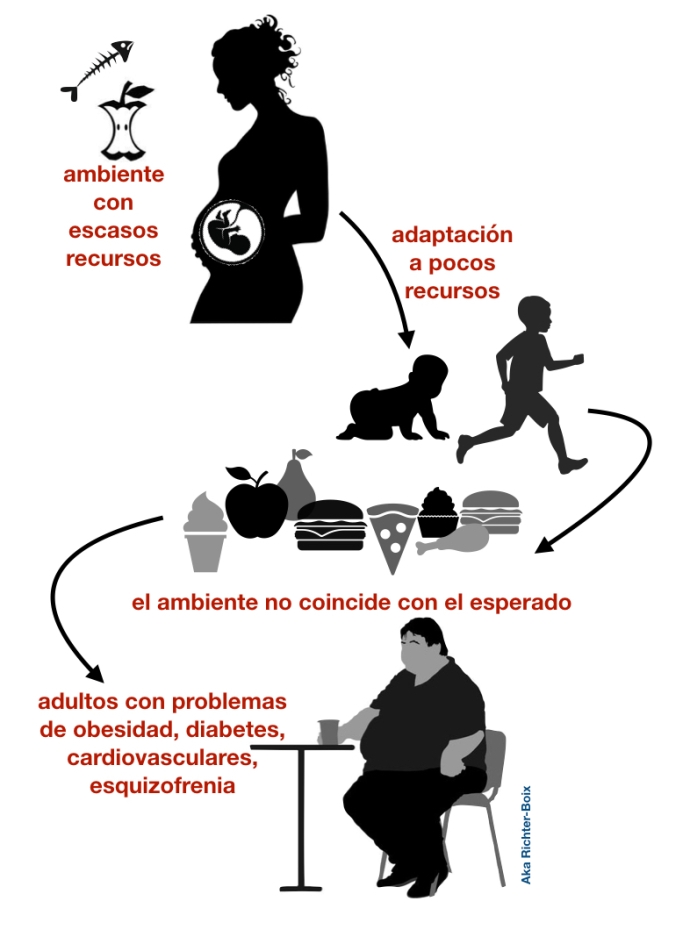


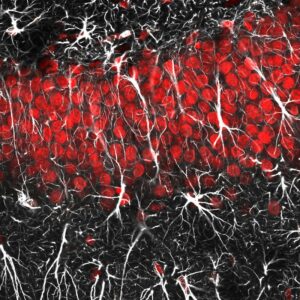


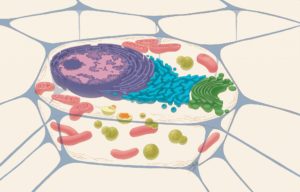



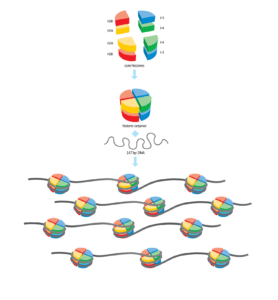

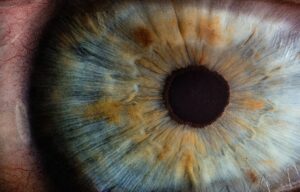






Debe estar conectado para enviar un comentario.