Recientemente se han descubierto una clase de ARN que podrían tener un importante papel en el desarrollo de algunos tipos de cáncer. Los cRNA destacan por su estabilidad, al poseer una estructura circular (obtenida mediante un proceso denominado back-splicing) que les protege de las ribonucleasas. El número de ARN circulares diferentes producidos en células humanas supera en gran proporción al número de ARN codificantes para proteínas, por lo que es lógico deducir que han de tener una importante función.
cRNA como herramientas diagnósticas
En ocasiones, los cRNA presentan patrones de expresión diferenciales en distintos tejidos o estadíos celulares, incluyendo estados patológicos como el cáncer. Por ejemplo, los rt-circRNAs aparecen cuando se da una transcripción defectuosa, en la que no se ha producido la terminación correctamente y se ha transcrito parte del gen contiguo, llevando a un procesamiento del ARN diferencial y apareciendo una molécula circular. La transcripción descontrolada que da lugar a estas situaciones ha sido relacionada con el cáncer.
Figura 1. Mecanismos de formación de ARN circular. A la derecha observamos la formación de rt-circRNA, tras la transcripción de dos exones de diferentes genes.
Gracias a esta expresión diferencial, podrían suponer una interesante herramienta diagnóstica y pronóstica. También se han relacionado cRNA con algunas características clínicas como el tipo de cáncer o su agresividad. Además, suponen una ventaja respecto a los diagnósticos convencionales, y es que gracias a esta estabilidad se podrían detectar en el plasma, la saliva o la orina.
cRNA como dianas terapéuticas
Los cRNA pueden tener numerosas funciones celulares, incluídas algunas que influyen en la pluripotencalidad celular, por lo que han sido relacionados con la formación de tumores. Por ejemplo, se ha demostrado que circFECR1 participa en la activación de un oncogén que favorece la metástasis en el cáncer de mama.
Figura 2. Los cRNA pueden ser reguladores de la transcripción (1); “esponjas” para los micro RNA, afectando a los genes de manera post-transcripcional (2); en ocasiones pueden traducirse (3); o modificar la estabilidad de otras moléculas de ARN (4), entre otras numerosas funciones celulares.
Es por esto que se ha planteado que la modificación de la expresión de algunos cRNA podría frenar la producción de tumores o la metástasis. Diversos métodos que los toman como posibles dianas terapéuticas están actualmente en investigación:
Por una parte, se está estudiando su silenciamiento por ribointerferencia.
Por otro lado, también se investiga el inducir su sobreexpresión, y utilizar su capacidad de actuar como “esponja” que limite la acción de miRNA oncogénicos.
Por último, también se han encontrado circRNA responsables de algunas resistencias terapéuticas. Un ejemplo es el circRNA-SORE. Tras aplicar en ratones un tratamiento que lo toma como diana, la respuesta al medicamento antineoplásico sorafenib fue restituida.
Figura 3. Resumen de las posibles aplicaciones de los cRNA en diversos tipos de cáncer: en color negro los posibles marcadores diagnósticos y pronósticos, y en azul las posibles dianas terapéuticas.
En conclusión, el descubrimiento de los circRNA ha abierto numerosas posibilidades para la investigación en cáncer, desde mejores herramientas diagnósticas a posibles vías de tratamiento. Aunque todavía existen limitaciones al no conocerse el completo la biogénesis y las funciones biológicas de estas moléculas, el futuro de estas líneas sin duda es muy prometedor.
Bibliografía
G. Pisignano, D. C. Michael, T. H. Visal, R. Pirlog, M. Ladomery, y G. A. Calin, «Going circular: history, present, and future of circRNAs in cancer», Oncogene, vol. 42, n.o 38, pp. 2783-2800, sep. 2023, doi: 10.1038/s41388-023-02780-w.
L. S. Kristensen, T. Jakobsen, H. Hager, y J. Kjems, «The emerging roles of circRNAs in cancer and oncology», Nat. Rev. Clin. Oncol., vol. 19, n.o 3, pp. 188-206, mar. 2022, doi: 10.1038/s41571-021-00585-y.
Se trata de medicina personalizada que se caracteriza por apuntar específicamente a ciertos cambios o sustancias presentes en las células cancerosas. Esto es lo que hace la tecnología de secuenciación masiva de alteraciones genómicas oncogénicas en tumores sólidos.
Imagen del equipo multidisciplinar implicado en las nuevas técnicas Gobierno de Aragón
Desde octubre de 2023, el Hospital Clínico de Zaragoza incorpora una técnica de análisis genómico exhaustivo sobre muestras de tumor a determinados pacientes oncológicos gracias a los avances en la secuenciación del genoma, especialmente con la implementación de técnicas de secuenciación de nueva generación (NGS), que permiten analizar el ADN tumoral de manera exhaustiva para poder hacer un diagnóstico más preciso del tumor y, en consecuencia, poder ofrecer un abordaje terapéutico más personalizado.
Después de una biopsia o cirugía, los tumores se someten a análisis para identificar alteraciones moleculares. Las técnicas de NGS facilitan el diagnóstico al analizar varios genes y alteraciones moleculares de manera simultánea y con un plazo de tiempo de entrega de resultados entre cinco o seis días.
Esto es de gran utilidad ya que, gracias al análisis de los resultados moleculares del tumor y las características específicas del paciente, se pueden tomar decisiones terapéuticas personalizadas y de seguimiento de la evolución de la enfermedad.
En un estudio publicado en un artículo científico de Current Biology se muestra como el ADN de un pelo de Beethoven ha podido desvelar la posible causa de su repentina muerte. Falleció en 1827 tras estar en cama muchos meses. Este gran músico se caracterizó por su sordera, que empezó a los 20, perdiendo la audición total a los 40. Con el estudio los investigadores quisieron descubrir más acerca de su salud. Para ello analizaron los genes procedentes de su cabello. Esto descartó que su muerte fuese por plomo, pensado anteriormente. Y aunque no pudieron analizar mucho del porqué de su sordera, sí descubrieron una enfermedad muy grave que padecía y que por tanto podía ser la causa de su muerte, la Hepatitis B. Sin duda, llama la atención como a partir de un pelo de hace casi 200 años se haya podido descubrir la enfermedad que sufría la persona del pasado. Gracias a todos los avances en tecnología y ciencia, unidos a los conocimientos de genética nos permiten avanzar hacia el futuro pero además, conocer mucho más sobre el pasado.
El hecho de que las mujeres representen aproximadamente el 80% de todos los casos de enfermedades autoinmunes como la artritis reumatoide o el lupus eritematoso sistémico (LES), ha llamado la atención a inmunólogos y reumatólogos durante los últimos años.
Prevalencia de dos enfermedades autoinmunes. La parte gris representa a mujeres y la blanca a hombres.
Las mujeres y los hombres sanos difieren en la composición y función de las células inmunitarias a nivel innato y adaptativo. Además, ellas tienden a desarrollar una respuesta inmune más robusta y a sufrir más enfermedades autoinmunes, especialmente las de tipo reumático. Estas disparidades se atribuyen a diferencias en las hormonas sexuales y en los genes del cromosoma X. La importancia de los genes del cromosoma X se ejemplifica en los hombres con síndrome de Klinefelter (47, XXY), que tienen un cromosoma X adicional y desarrollan LES con tanta frecuencia como las mujeres.
¿Hormonas o cromosoma X?
La mayor incidencia de enfermedades reumáticas autoinmunes durante los años reproductivos sugiere que las hormonas sexuales están implicadas. Un ejemplo de ello es la proporción entre mujeres y hombres con LES que antes de la pubertad es de 2:1, y después de 9:1. En general, los estrógenos tienen efectos inmunoestimulantes sobre el sistema inmunológico adaptativo, mientras que los andrógenos tienen efectos inmunosupresores.
Aunque numerosos estudios han evidenciado el papel de la señalización de los receptores de hormonas sexuales, especialmente los estrógenos, estos no son la única causa de la predisposición femenina a la autoinmunidad. El cromosoma X y su casi inactivación como forma de compensación génica entre machos y hembras se han puesto en el punto de mira.
Asimismo, las hormonas sexuales pueden inducir cambios epigenéticos. Por ejemplo, los estrógenos, al regular negativamente la expresión de la ADN metiltransferasa 1 la cual conduce a la hipometilación del ADN en las células T de pacientes femeninas con LES, pueden contribuir a la autoinmunidad en esta enfermedad.
Compensación génica e inactivación del cromosoma X
Debido a que las mujeres tienen dos cromosomas X y los hombres tan solo uno, la regulación de la dosis de los genes ligados al cromosoma X es estrictamente necesaria. El aumento anormal de la expresión de genes ligados al cromosoma X conduce a una disfunción del desarrollo. Por ello, en las hembras de los mamíferos uno de los dos cromosomas X se inactiva aleatoriamente en el embrión temprano para equilibrar la dosis de expresión genética entre los sexos. El proceso de inactivación del cromosoma X (XCI) da como resultado un mosaicismo celular. Sin embargo, las regiones pseudoautosómicas (PAR) del cromosoma X, PAR1 y PAR2, no se someten a XCI. Los genes de esta región tienen homólogos en el cromosoma Y.
No obstante, la inactivación del cromosoma X (XCI) no es completa y entre el 15 y el 23 % de los genes del cromosoma X inactivo (Xi) escapan del XCI, contribuyendo así a la aparición de una población heterogénea de células femeninas específicas.
El proceso de inactivación es iniciado por el largo ARN no codificante XIST (verde), que se expresa altamente en un alelo y recubre el futuro cromosoma X inactivo (Xi) en cis , lo que lleva a la represión transcripcional. El resultado es un mosaico celular donde en teoría la mitad de las células tienen un cromosoma activo (Xa) de origen materno y la otra mitad de origen paterno ( B ). Sin embargo, ciertos genes pueden escapar de XCI ( C ).
Algunos genes relacionados con el sistema inmunitario pueden escapar de esta inactivación en grado y penetrancia variables, lo que da como resultado una expresión bialélica en algunas células inmunitarias, como TLR7. Debido a que es necesaria una regulación estricta de la expresión de TLR para un entorno inmunológico saludable y autotolerante, se ha propuesto el escape de XCI como un mecanismo que contribuye a este dimorfismo sexual.
Genes ligados al cromosoma X que escapan de XCI contribuyendo a las enfermedades autoinmunes
Los genes ubicados fuera de las regiones pseudoautosómicas codifican proteínas y microARN que juegan un papel directo o indirecto en la regulación de la respuesta inmune. Mutaciones que resultan en la pérdida de la función de algunos de estos genes como BTK, WAS, CXCR 3 y CD 40L pueden causar deficiencias inmunes primarias.
Se sabe que el cromosoma X contiene la mayor cantidad de genes relacionados con el sistema inmunológico de todo el genoma humano.
Entre los genes más importantes se encuentran los receptores endosómicos tipo Toll (TLR), específicamente TLR7 y TLR8. Estos genes desempeñan funciones vitales no redundantes. TLR7 tiene un gran papel en ciertas infecciones al identificarse, por ejemplo, mutaciones de pérdida de su función asociadas a formas graves de COVID-19 en individuos jóvenes.
Cromosoma Y e implicaciones en el sistema inmunológico
El cromosoma Y es uno de los cromosomas más cortos del genoma humano, es heterocromático y está principalmente compuesto por genes multicopia, secuencias repetidas y elementos transponibles.
Además de poseer el gen SRY, contiene un total de 64 genes codificadores de proteínas con un papel activo en la regulación genética, afectando a la dinámica de la cromatina a través de sus genes ribosómicos multicopia.
Ciertas variaciones genéticas como la pérdida en mosaico de cromosomas Y (LOY) pueden tener efectos sobre la función inmunológica pudiendo afectar a la vía IMD (Immune Deficiency) y a la susceptibilidad a enfermedades autoinmunes, como es el caso de la encefalomielitis autoinmune experimental.
Además, se ha encontrado una relación entre LOY y una menor expresión de ciertos genes inmunológicamente relevantes en los leucocitos de individuos masculinos, lo que podría tener implicaciones en la función inmunológica general.
Conclusión
El predominio de mujeres con ciertas enfermedades autoinmunes sugiere una influencia de factores biológicos como las hormonas sexuales y la genética, especialmente el cromosoma X. Mientras que las hormonas sexuales, como los estrógenos, pueden tener efectos inmunoestimulantes, los andrógenos muestran efectos inmunosupresores. Asimismo, el cromosoma X contiene numerosos genes relacionados con la respuesta inmunitaria por lo que una sobreexpresión de alguno de estos genes debido al escape de la inactivación de uno de los dos cromosomas X, puede alterar la respuesta inmunitaria apareciendo así enfermedades autoinmunes.
Por último, el cromosoma Y también muestra implicaciones en el sistema inmunológico por su papel en la regulación genética.
El plegamiento incorrecto grave del ADN y el silenciamiento de genes están relacionados con el tejido sináptico y conectivo vinculado al síndrome de X frágil.
Investigadores de la Universidad de Pennsylvania revelan que la mutación responsable del síndrome del X frágil altera la expresión de diferentes genes relacionados con la función nerviosa y la integridad del tejido conectivo. También muestran una estrategia para restaurar la expresión de estos genes, mediante edición genómica.
El síndrome del X frágil es un trastorno genético que afecta al cromosoma X. Produce problemas en el desarrollo, en el aprendizaje y deterioro cognitivo. Este síndrome representa la causa más frecuente de discapacidad intelectual hereditaria y afecta con mayor frecuencia y gravedad a los hombres.. Aunque no tiene cura, existen diferentes aproximaciones terapéuticas.
Este síndrome está causado por una alteración en el gen FMR1, que codifica para la proteína FMRP (ribonucleoproteína mensajera del X frágil), la cual regula la síntesis de proteínas y otras rutas de señalización en las células nerviosas.
La alteración es producida por una mutación denominada “expansión repetida”, la cual indica la repetición de una secuencia de ADN que se vuelve larga e inestable. En este caso, la expansión de nucleótidos en la región del gen FMR1 de una persona sana son 40 repeticiones del trinucleótido CGG. Pero según el grado de afección de la enfermedad se puede encontrar entre 60 y 200, siendo este último el más grave. Esta anomalía desencadena una respuesta defensiva de la célula, silenciando a FMR1 y FMRP .
El estudio en animales pequeños impedía ver la expansión de ADN, por lo que estos estudios recientes tienen implicaciones para las futuras estrategias de tratamiento del síndrome X frágil y ponen en relieve los posibles mecanismos que contribuyen a la inestabilidad del genoma que pueden subyacer a otras enfermedades también.
Inactivación de la cromatina e inestabilidad genómica
Los estudios de esta universidad se centraron en estudiar con detenimiento el impacto de la expansión del ADN en la enfermedad mediante técnicas avanzadas de secuenciación e imágenes. A partir de células de pacientes, tejidos muertos y diversos tipos celulares, los investigadores han encontrado evidencias de que el cambio de conformación del ADN que genera la expansión CGG altera la funcionalidad del genoma más allá de FMR1, todo debido a la metilación y heterocromatización.
Los investigadores descubrieron que grandes franjas de múltiples cromosomas en muestras de pacientes con FXS que incluyen la repetición CGG, están marcadas con la heterocromatina silenciadora. Estos dominios de heterocromatina se denomina BREACHes: balizas de expansión repetida, anclaje y contacto con heterocromatina.
Las BREACH se unen en grupos que entran en contacto físico en el núcleo y silencian genes implicados en las conexiones sinápticas de las neuronas, junto con genes vinculados a la integridad del tejido conectivo, como la piel y las articulaciones.
Las alteraciones producen un mal plegamiento de la cromatina de las regiones adyacentes a FMR1 y el acercamiento físico entre la región FMR1 y las de otros cromosomas también inactivados en pacientes con la enfermedad.
Como consecuencia de todo esto, se silencia la expresión de numerosos genes implicados en la plasticidad sináptica, la adhesión neural, desarrollo testicular.
Conclusiones de la investigación y vías terapéuticas a explorar
Este estudio ha demostrado que esta enfermedad va más allá de un trastorno monogenético, en el que es afectado un gen, sino que intervienen otros mecanismos que afectan a la conformación y función de otras regiones cromosómicas.
Se están buscando diferentes vías de acción terapéutica. Entre ellas queda pendiente el estudio de los genes de otros cromosomas cuya expresión se ve alterada, siendo estos posibles dianas de los tratamientos.
Los investigadores probaron si la repetición estaba relacionada con las BREACHes mediante el uso de técnicas como CRISPR-Cas, con el objetivo de reducir la longitud de la expansión.
Gracias a esto, acortaron la expansión de CGG a longitudes menores (100-190). Esto condujo a muchas de las franjas de heterocromatina silenciadoras a invertirse y por ende, a muchos de los cromosomas a desconectarse de FMR1. De esta forma, FMR1 no interactuaba con otras regiones envitando así que se silenciaran. Los experimentos del equipo demostraron que los genes originalmente silenciados por BREACH se reexpresaron en células FXS con la repetición CGG acortada por CRISPR.
Los investigadores establecieron como estrategia ambiciosa el reducir la expansión excesiva de las repeticiones de CGG en un momento del desarrollo para prevenir o mitigar los efectos del silenciamiento de la heterocromatina. Para explorar esta posibilidad sería necesario equilibrar cuidadosamente los efectos positivos de la reactivación de genes importantes con el papel protector que tiene la heterocromatina contra la inestabilidad del genoma repetitivo.
Ejemplos de otros trastornos potencialmente afectados por estos hallazgos incluyen la enfermedad de Huntington y la esclerosis lateral amiotrófica (enfermedad de Lou Gehrig), que son miembros de la misma clase de trastornos de expansión repetida que el FXS.
Los resultados sugieren que es posible que en el futuro se descubra que las BREACHes tienen un impacto más amplio en el silenciamiento de genes en otras enfermedades con inestabilidad genómica, incluidos ciertos cánceres y otros trastornos de expansión repetida.
El 15 de diciembre de 2023 fue aprobada por parte de la EMA (Agencia Europea del Medicamento) la autorización para la comercialización del primer tratamiento basado en la modificación del genoma con la técnica de edición genética CRISPR. Se trata de un avance médico que se podrá emplear en pacientes mayores de 12 años que padezcan anemiafalciforme. Esta agencia fue la tercera en realizar esta aprobación, puesto que, en los quince días anteriores, ya lo habían hecho tanto la agencia británica como la estadounidense.
¿Qué es la anemia falciforme?
La anemia falciforme es una enfermedad debilitante y potencialmente mortal que pertenece a un grupo de trastornos hereditarios que afectan a los glóbulos rojos. Los glóbulos rojos o eritrocitos son los elementos formes encargados del transporte de oxígeno a través de la sangre. Esto es posible gracias a que contienen un pigmento denominado hemoglobina.
Los eritrocitos sanos son redondos, con forma de disco, y sus paredes son lo suficientemente flexibles para que puedan adaptar su forma según avanzan por los diferentes vasos sanguíneos del cuerpo. En el caso de la anemia falciforme, estas células adoptan forma de hoz, y pierden esa elasticidad en sus paredes. Esto hace que se puedan quedar acumulados en los vasos sanguíneos, formando tapones que bloquean estos vasos, causando dolores que son incompatibles con la vida. Además, se suelen producir otras muchas complicaciones debido a que los coágulos evitan que el riego sanguíneo llegue a determinadas zonas del organismo.
La anemia falciforme es una enfermedad hereditaria, causada por una mutación en el gen HBB, localizado en el cromosoma 11, en su brazo largo. Este gen es el encargado de la producción de la subunidad beta de la hemoglobina, que hemos dicho que es la proteína encargada del transporte de oxígeno en la sangre. La hemoglobina está formada por cuatro subunidades: dos alfa y dos beta. La subunidad beta está formada por 8 hélices alfa, además de poseer un grupo hemo que le permite unirse al oxígeno.
Por tanto, mutaciones en este gen pueden provocar graves alteraciones en la funcionalidad de la hemoglobina.
La mutación se produce en el sexto aminoácido que forma la subunidad, intercambiándose el glutamato por la valina. El glutamato tiene carga negativa, mientras que la valina no, no es polar, por lo que la hemoglobina va a tener dos cargas negativas menos (una por cada subunidad beta mutada), haciendo que se reduzca la solubilidad. Además, la valina es hidrofóbica, lo que genera puentes con alanina, fenilalanina y leucina, creando polímeros dentro de la estructura celular. Cuando la presión de oxígeno baja, la hemoglobina mutada va a cristalizar.
Todo en conjunto va a hacer que el eritrocito adquiera esa forma alterada característica de la enfermedad. Cuando la hemoglobina está mutada, se denota como hemoglobina S, pero cuando está en estado normal se denomina hemoglobina A.
La forma de herencia es autosómica recesiva. Se requieren dos copias del gen mutado para que tenga lugar la enfermedad en su totalidad. Esto hace que tengamos dos formas de desarrollo de la enfermedad, en función de cuantas copias se hereden:
Anemia falciforme: es el desarrollo completo de la patología, por lo que se presenta cuando el individuo es homocigoto recesivo para la forma mutada. Es la forma más grave, y tiene dos consecuencias principales: la vasoconstricción y la anemia hemolítica. La segunda es la destrucción de los eritrocitos, y la primera es la consecuencia de la acumulación de los eritrocitos irregulares en los vasos sanguíneos.
Rasgo falciforme: se presenta cuando el individuo es heterocigoto para el gen mutado, de manera que solamente presenta una copia. En este caso, solo es portador de la enfermedad, y la cantidad de hemoglobina S, errónea, que se genera es muy inferior a la cantidad de hemoglobina A. Estas personas pueden llevar una vida totalmente normal, aunque podrían llegar a presentar problemas si realizan actividades físicas muy extremas.
¿En qué se basa la técnica CRISPR?
La técnica CRISPR/Cas fue descubierta por un grupo de investigación de la Universidad de Osaka al estudiar la resistencia de algunas bacterias y arqueas ante diversos fagos. En este caso, se vio que en Escherichia coli había zonas del genoma con secuencias de nucleótidos repetidas, altamente conservadas. Primero se pensó que era ADN basura, pero más adelante se vio que las secuencias localizadas entre esas zonas de repetición, denominas zonas espaciadoras, eran complementarias a las secuencias de algunos fagos y virus patógenos de estas bacterias estudiadas.
Los investigadores Koonin y Makarova propusieron que las bacterias integraban fragmentos del genoma de los fagos en su propio material genético como parte de un “sistema inmune”, de manera que, al ser transcritas, cuando entre otra vez el mismo fago van a poder reconocer su genoma y eliminarlo, deteniendo el proceso infectivo. Estas secuencias pasaron a denominarse CRISPR, y se pasó a estudiar su aplicación en edición genómica junto a proteínas Cas, que funcionan como las verdaderas “tijeras moleculares”, ya que pueden cortar cualquier secuencia de nucleótidos en un punto concreto al actuar como endonucleasas.
El complejo CRISPR/Cas es por tanto el mecanismo de defensa en bacterias y arqueas. Está formado por dos elementos: las secuencias CRISPR, que son trozos de ARNcr, y la endonucleasa Cas. La primera es la que reconoce las secuencias complementarias e indica a las proteínas Cas donde tienen que realizar el corte. Gracias a la precisión de los cortes que puede realizar la proteína Cas este mecanismo es muy útil a la hora de realizar ediciones en el genoma.
El objetivo original es su uso para el silenciamiento de determinadas secuencias, al eliminar cierta zona del material genético al recortarlo, aunque puede usarse para la adición de secuencias o la inserción de mutaciones.
Aplicación de CRISPR/Cas para la anemia falciforme
El tratamiento, que ha sido llamado Casgevy, se recomienda para aquellos pacientes mayores de 12 años cuyo tratamiento ideal sería un transplante de células madre hematopoyéticas sanas, pero este no se puede llevar a cabo porque no hay un donante adecuado disponible. El transplante de médula ósea es el tratamiento habitual, y puede llegar a curar la enfermedad, aunque también se pueden llevar a cabo transfusiones sanguíneas, lo que solamente ayuda a disminuir los síntomas y las complicaciones, sin llegar a eliminar el problema.
Con esta terapia de edición genética, se extraen células madre del propio paciente, y son estas mismas células las que se modifican e inyectan de nuevo al individuo, lo que hace que sea una técnica mucho más especializada. Además, se reducen las posibilidades de rechazo al ser un autotransplante.
El procedimiento que se sigue con el tratamiento consiste en el silenciamiento de la zona mutada del gen HBB para que cese la producción de beta globina. Esta subunidad es sustituida por la gamma globina, que forma parte de la hemoglobina fetal de manera que la hemoglobina puede volver a ser funcional. Esta hemoglobina fetal es eliminada casi en su totalidad a lo lago de los primeros años de vida, pero al dejar de producir la beta globina, se reactiva su formación.
Las células modificas son introducidas de nuevo en los pacientes, de manera que estos pueden desarrollar sus propios eritrocitos funcionales, reduciendo significativamente los síntomas que causa la enfermedad.
Conclusiones
La aprobación por parte de las tres agencias de este tratamiento es un avance muy importante en el campo de la edición genética y una buena noticia en el ámbito de la medicina, ya que existen escasas terapias para este tipo de enfermedades. El problema reside en que se calcula que puede llegar a costar unos 2 millones de dólares por paciente, lo que hace que esté al alcance de muy pocos llegar a recibir este tratamiento.
Bibliografía
Esrick EB, et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. NEJM. 2020
Giono, L. E. (2017). CRISPR/Cas9 y la terapia génica. MEDICINA (Buenos Aires), 77(5), 405-409.
Lammoglia-Cobo, M. F., Lozano-Reyes, R., García-Sandoval, C. D., Avilez-Bahena, C. M., Trejo-Reveles, V., Muñoz-Soto, R. B., & López-Camacho, C. (2016). La revolución en ingeniería genética: sistema CRISPR/Cas. Investigación en discapacidad, 5(2), 116-128.
La gripe aviar es responsable de entre 3 y 5 millones de casos graves en aves y de entre 250 y 500.000 muertes al año. Aunque la mayoría de las cepas solo afectan a las aves, algunas pueden afectar a los humanos. Desde 2022 ha habido un aumento en los casos de gripe aviar en el ser humano lo que podría llegar a causar una pandemia. Para evitar que la enfermedad se extienda y ser contagiados, existen dos opciones: modificar al ave para que no se infecte, y por tanto no infectar a los humanos, o modificar al humano para que no se contagie del ave infectada.
Modificaciones en aves La gripe aviar depende de la proteína ANP32A, en concreto de la subunidad PB2, para llevar a cabo sus funciones vitales. Se llevó a cabo un experimento que editó el gen ANP32A en células germinales de pollo con CRISP/Cas9. Las modificaciones llevadas a cabo fueron en 3 pares de bases del exón 4 que dan lugar a modificaciones en 2 aminoácidos.
Se descubrió que 9 de cada 10 pollos adultos eran resistentes al virus en bajas dosis. El problema venía cuando se empleaban dosis más altas porque el virus mutaba y en vez de depender de la proteína ANP32A, dependía de otras proteínas de la misma familia como ANP32, ANP32B o ANP32E.
Modificaciones en humanos Un estudio realizado por el MRC-University of Glasgow Center for Virus Research (CVR) identificó al gen BTN343 como la principal defensa por parte del ser humano contra la gripe aviar. Este gen BTN343 apareció por primera vez en primates, en concreto en el suborden Haplorrhini, como defensa contra virus. Se expresa principalmente en el epitelio nasal, bronquiolos, células alveolares de tipo I y II y macrófagos alveolares. La mayoría de las cepas de la gripe aviar no pueden atravesar sus defensas ya que bloquea la replicación y transcripción del virus. El problema es que cada vez hay más cepas resistentes a BTN343. El experimento llevado a cabo por el CVR consiguió identificar las mutaciones que hacen los hacen resistentes a BTN343. Pudiendo así identificar que virus podrían ser contagiados a las personas y cuales no, pero consiguieron modificar BTN343 para que también bloqueara al resto de cepas.
Conclusión La modificación en aves tiene un futuro más claro que en humanos, de eso no hay duda. No solo por los estudios mostrados, sino también por las facilidades que puede tener a la hora de realizar los experimentos. Igualmente se debería seguir trabajando en ambas direcciones, porque, aunque esta segunda tenga más dificultades, podría resultar muy útil, ya que no solo evitaríamos los contagios por gripe aviar, también muchas otras zoonosis.
Bibliografía LeMieux, J., PhD. (2023, 12 octubre). Avian flu target in chickens disguised by CRISPR. GEN – Genetic Engineering and Biotechnology News. https://www.genengnews.com/topics/infectious-diseases/avian-flu-target-in-chickens-disguised-by-crispr/# Identificado un gen clave para proteger a los humanos contra los virus de la gripe aviar | PortalVeterinaria. (s. f.). https://www.portalveterinaria.com/salud-publica/actualidad/41106/identificado-un-gen-clave-para-proteger-a-los-humanos-contra-los-virus-de-la-gripe-aviar.html Camacho-Zarco, AR, Kalayil, S., Maurin, D. et al. Base molecular de las interacciones huésped-adaptación entre la subunidad PB2 de la polimerasa del virus de la influenza y ANP32A. Nat Comuna 11 , 3656 (2020). https://doi.org/10.1038/s41467-020-17407-x Pinto, RM, Bakshi, S., Lytras, S. et al. La evasión de BTN3A3 promueve el potencial zoonótico de los virus de la influenza A. Naturaleza 619 , 338–347 (2023). https://doi.org/10.1038/s41586-023-06261-8
La espermatogénesis es el proceso por el cual las espermatogonias dan lugar a los espermatozoides, es decir, el proceso de diferenciación celular por el cual se producen los espermatozoides en el interior de los túbulos seminíferos. Este evento comienza poco antes de la pubertad bajo la influencia de concentraciones cada vez mayores de gonadotropinas hipofisarias, y continúa durante toda la vida.
Proceso de diferenciación celular de las espermatogonias indiferenciadas en espermatozoides.
Introducción
Un estudio reciente realizado por la Universidad de Washington, y publicado el 17 de abril de 2023 en la revista Nature Communications, establece que se ha identificado un gen común en mamíferos cuya función es la regulación del proceso de la espermatogénesis. Se trata del gen arrdc5.
Estos investigadores realizaron el estudio del transcriptoma de tejido testicular de diversos animales mamíferos como el ratón, el toro y el cerdo; además de en humanos. Con ello se destacó al gen arrdc5, cuya expresión parecía estar enriquecida.
Este gen codifica para la proteína ARRDC5 (Arrestin-Domain-Containing5). Se trata de una proteína de la familia de las arrestinas. A día de hoy solamente se han descubierto diez arrestinas en mamíferos: cuatro β-arrestinas y seis α-arrestinas (donde incluimos la proteína de interés). Se trata de una familia de proteínas desensibilizadoras de los receptores de proteína G, implicada esta última en la transducción de señales. El porqué de la implicación de ARRDC5 en la espermatogénesis es todavía incierta.
Proteína ARRDC5 en visión tridimensional.
¿Cómo se ha realizado el estudio?
Para la realización del estudio, que fue aprobado por el comité de uso y cuidado de los animales de la Universidad de Washington, se utilizaron diversos animales, como ratones, toros y cerdos.
Al inicio del proyecto se castraron ejemplares de estos animales para poder realizar diversos estudios. A partir de los testículos se realizaron varias técnicas para obtener células aisladas del tejido testicular. Con estas células se realizó posteriormente una digestión enzimática con tripsina-EDTA, colagenasa IV y DNasa I.
El siguiente paso realizado fue la amplificación por RT-PCR de los mRNA encontrados en las células testiculares. Además del tejido testicular, también se utilizaron otros tejidos para poder comparar los transcriptomas realizados. Entre estos tejidos se encuentran el cerebral, renal, hepático, cardiaco, adiposo, pancreático, pulmonar, muscular y del epidídimo.
La RT-PCR se realizó con 1µg de RNA y mediante el uso de primers específicos para amplificar el gen arrdc5. Estos primers se diseñaron exclusivamente para cada uno de los tres animales implicados en el proyecto a partir de sus genomas de referencia en las bases de datos. Tras la RT-PCR se realizó una electroforesis en gel de agarosa para observar los resultados de la amplificación.
Como se observa en la imagen adjunta, el gen arrdc5 se encuentra expresándose solamente en el tejido testicular de los tres animales implicados, y en ningún otro tejido. Por esta razón se llegó a la conclusión de que el gen solamente se expresaba en el tejido testicular y que en los demás tejidos del animal se encontraba inactivado.
Una vez se llega a esta conclusión se continúa con el proyecto. ¿Qué ocurre si producto del gen arrdc5 no está presente en ningún momento en los animales implicados?
Para salir de dudas se crearon ratones knockout para el gen arrdc5. Para ello se utilizó la técnica de edición por CRISPR-Cas9. En estos ratones knockout se produce una deleción de una porción bastante extensa del exón 1 que codifica para parte de la secuencia del gen arrdc5. Con ello, los ratones presentarán mutado el gen (gran parte de este ha sido delecionado). Las proteínas, por tanto, que codifican el gen arrdc5 no serán funcionales.
Para estudiar cómo afecta este gen a la fertilidad de los animales se aparearon ratones arrdc5-/- con ratonas salvajes; además de ratonas arrdc5-/- con ratones silvestres (como control).
Acción de CRISPR-Cas9 sobre el exón 1 del gen arrdc5 y sus consecuencias
Conclusiones
Tras todos los procedimientos realizados, estudios histológicos, inmunohistoquímicos, genéticos y analíticos se llegó a la conclusión de que el gen arrdc5 estaba implicado en la espermatogénesis en machos. Es un gen esencial en el proceso y cuya carencia induce a los animales en presentar deficiencias para generar el número óptimo de espermatozoides, una menor movilidad de los mismos y anormalidades estructurales.
Todas estas características hacen que los machos sean infértiles y sufran de oligoastenoteratospermia. Esta condición de infertilidad se ha podido encontrar en las especies de mamíferos, ganado doméstico, especies salvajes e incluso en humanos. Por esa razón se sabe que los mecanismos que regulan la óptima espermatogénesis se han conservado a lo largo de la evolución. En cambio, los procesos moleculares que se producen en los ejemplares infértiles son todavía inciertos.
¿Qué se busca para el futuro?
Muchos hombres suelen recurrir a la vasectomía, cirugía por la cual se realizan unas incisiones en los conductos deferentes para evitar la salida de los espermatozoides desde los testículos a la uretra. Se trata de un método anticonceptivo permanente con una efectividad del 99%.
Dibujo ilustrativo de la vasectomía
Con las conclusiones obtenidas de este estudio se busca la posibilidad de poder lanzar al mercado un nuevo fármaco anticonceptivo para hombres que no sea permanente. Se trataría de un inhibidor temporal de la expresión del gen arrdc5. Por tanto, los hombres que tomen este fármaco se volverían estériles temporalmente, durante el tiempo de acción del inhibidor.
Con este nuevo fármaco se quiere evitar el uso de anticonceptivos hormonales, que, aunque son útiles, presentan efectos secundarios. La testosterona puede producir una supresión en la formación de espermatozoides, permitiendo por tanto la azoospermia. En cambio, esta hormona también está implicada en otros procesos fisiológicos de gran importancia como las funciones cognitivas (memoria, concentración, …), prevención de problemas cardiovasculares, regulación de la densidad ósea y muscular, … Por ello se piensa que este nuevo fármaco sería de gran utilidad puesto que actúa sobre una diana específica y no afecta a otras funciones del organismo.
Bibliografía
ARRDC5 expression is conserved in mammalian testes and required for normal sperm morphogenesis
La cromotripsis es un mecanismo mutacional que da lugar a reordenamientos genómicos que afectan a un cromosoma o a una pequeña cantidad de estos. La palabra proviene del griego chromo de cromosoma y thripsis de «romperse en pedazos».
Este fenómeno es especialmente importante en el estudio del cáncer, sin embargo, hoy hablaremos de cómo este mecanismo logró curar el síndrome de WHIM.
Más cosas sobre el síndrome de WHIM
El síndrome de WHIM es una inmunodeficiencia primaria de herencia autosómica dominante, que produce verrugas, hipogammaglobulinemia, (concentración baja de inmunoglobulinas) infecciones bacterianas recurrentes y mielocatesis (cuadro clínico caracterizado por neutropenia crónica).
Imagen 1. Verrugas pequeñas distribuidas en dorso de mano. Se observan lesiones características agrupadas en nudillos
Esta enfermedad está relacionada con el gen CXCR4, que codifica para una proteína que se localiza en la superficie celular y que se encarga de reconocer quimiocinas. En los pacientes con WHIM, una de las copias del gen funciona con normalidad, pero la otra es defectuosa, dando lugar a una hiperactividad que hace que los glóbulos blancos permanezcan “atrapados” en la médula ósea en lugar de transportarse a la sangre.
Caso real
El caso de hoy es de una mujer nacida en los años 50 que padecía esta enfermedad. Aproximadamente 60 años después, contactó con servicios médicos para confirmar que sus dos hijas habían heredado dicha enfermedad. Cuando se evaluó a sus hijas, se preguntó a la paciente como se sentía y ella confirmó no haber presentado ningún síntoma desde hacía 30 años. Además, sus glóbulos blancos no presentaban la mutación del gen CXCR4 antes mencionada.
Tras esto, se analizó su cariotipo y se encontró que una copia del cromosoma 2 era un 15% más corta que la otra, lo que significaba que el fragmento delecionado era el causante de la enfermedad.
Es por esto que se llegó a la conclusión de que la cromotripsis se había encargado de curar la enfermedad de esta paciente, aunque en este caso ocurrió en una célula madre sanguínea, lo que dio lugar a nuevas células sanguíneas “sanas”.
Imagen 2 (National Institute of Allergy and Infectious Diseases, NIH).Cariotipo de una mujer curada espontáneamente del síndrome WHIM. Estos pares de cromosomas, que son de sus glóbulos blancos, muestran un cromosoma 2 normal a la izquierda y un cromosoma 2 truncado a la derecha.
¿Cómo funciona?
Tras haber hablado de este fascinante caso, lo que de verdad importa es saber cómo se ha producido esta alteración cromosómica.
Imagen 3 (Nazaryan-Petersen, L., Bjerregaard, V. A., Nielsen, F. C., Tommerup, N., & Tümer, Z. (2020). Chromothripsis and DNA Repair Disorders. Journal of clinical medicine). Roturas de doble cadena (DSB) y mecanismos de reparación.
Este mecanismo se caracteriza por la “destrucción” localizada de ADN o por la formación de bucles, así como la posterior reunión de los fragmentos. Es durante el reensamblaje cuando se producen las deleciones.
Los fragmentos se pueden reparar a través de distintos mecanismos de reparación de ADN. Las dos formas principales son la recombinación homóloga (sin errores), y la unión de extremos no homólogos (propensa a errores)
Lo más posible es que la unión de estos fragmentos no homólogos se produzca gracias a los mecanismos de reparación de ADN que actúan de forma natural en las células.
Conclusión
Los expertos afirman que este caso confirma que la cromotripsis puede tener efectos curativos, por lo que es un mecanismo que debería ser estudiado en otras enfermedades genéticas.
Bibliografía
Kaiser, J. Shattered chromosome cures woman of immune disease (2015). doi: 10.1126/science.aaa7807
Dr. Collins, F. Shattering News: How chromotrypsis cured a rare disease (2015).
Nazaryan-Petersen, L., Bjerregaard, V. A., Nielsen, F. C., Tommerup, N., & Tümer, Z. (2020). Chromothripsis and DNA Repair Disorders. Journal of clinical medicine, 9(3), 613. https://doi.org/10.3390/jcm9030613
Alrededor de un 17% de las mujeres con infertilidad sin causa conocida son portadoras de variantes genéticas responsables de causar enfermedades, concluye un reciente estudio dirigido por investigadores de la Universidad de Augusta (EE.UU) y publicado en New England Journal of Medicine.
En aproximadamente un 30% de las mujeres con problemas de infertilidad se desconoce cuál es la causa de que no puedan quedarse embarazadas por ellos los investigadores de la universidad ya mencionada analizaron el exoma de 197 mujeres con infertilidad de causa desconocida y lo comparó con el de la población general.
El equipo detectó que 13 de las mujeres con infertilidad analizadas eran portadoras de variantes en genes accionables, esto es en genes responsables de condiciones para las que existen opciones terapéuticas o preventivas. Estas variantes identificadas estaban relacionadas con enfermedad cardiovascular o cáncer. Por ejemplo, cuatro mujeres eran portadoras de variantes en BRCA1 o BRCA2 y una era portadora de una variante del gen MYH11 asociada a un riesgo elevado a desarrollar una rotura aórtica. En población general este tipo de variantes se detectó en menos del 2% de las personas analizadas.
Además en 21 de las mujeres con infertilidad de causa determinada los investigadores detectaron variantes genéticas responsables de causar enfermedades como la esclerosis lateral amiotrófica, enfermedad renal poliquística o Parkinson, para las que existen menos oportunidades terapéuticas.
De momento no se ha establecido una conexión molecular directa entre la infertilidad y las enfermedades causadas por las variantes identificadas. De hecho, una cuestión pendiente será evaluar si las variantes identificadas pueden estar implicadas de forma simultánea en la infertilidad y la enfermedad correspondiente. También deberán replicarse los resultados en una muestra de mayor tamaño.
Bibliografía.
Dougherty MP, et al. Unexplained Female Infertility Associated with Genetic Disease Variants. N Engl J Med. 2023 Mar 16;388(11):1055-1056
La noticia fue publicada el día 12 de mayo de 2023.
La impactante noticia afirma que un bebé ha sido fecundado con ADN de tres personas con el objetivo de evitar que los niños hereden enfermedades incurables.
La mayor parte del ADN del bebé proviene de su padre y su madre biológicos, y alrededor del 0,2% de una tercera mujer donante.
Para ello han usado una técnica conocida como tratamiento de donación mitocondrial(MDT, por sus siglas en inglés) utiliza tejido de los óvulos de mujeres donantes sanas para crear embriones en Fecundación In Vitro (FIV) libres de mutaciones dañinas que portan sus madres y que es probable que se las transmitan a sus hijos. Las enfermedades mitocondriales son incurables y pueden ser fatales a los pocos días o incluso horas del nacimiento.
Como cualquier otra persona, los bebés concebidos a través de este método nacen de la fertilización de un solo espermatozoide y un solo óvulo. La diferencia radica en la relación entre el material nuclear que alberga la gran mayoría de los genes responsables de construir un ser humano y la pequeña cantidad de ADN no nuclear que ayuda a fabricar las unidades de energía de la célula, llamadas mitocondrias.
Debido a que los embriones combinan el esperma y el óvulo de los padres biológicos con las diminutas estructuras mitocondriales del óvulo de la donante, el bebé resultante tiene ADN de la madre y el padre, además de una pequeña cantidad de material genético (alrededor de 37 genes) de la donante.
Aunque hay presencia del ADN de una donante, el 99,8% del ADN del recién nacido procede de la madre y el padre.
Las personas heredan mitocondrias de su madre, por lo que las mutaciones dañinas pueden afectar a todos los hijos. Las mitocondrias son los pequeños compartimentos dentro de casi todas las células del cuerpo que convierten los alimentos en energía utilizable, cuando son defectuosas no alimentan el cuerpo y provocan daño cerebral, atrofia muscular, insuficiencia cardíaca y ceguera.
Para las mujeres afectadas, la concepción natural suele ser una incertidumbre, ya que algunos bebés pueden nacer sanos porque heredan solo una pequeña proporción de las mitocondrias mutadas, pero otros pueden heredar mucho más y desarrollar enfermedades graves, progresivas y, con frecuencia, mortales.
La investigación sobre tratamiento de donación mitocondrial, fue iniciada en el Reino Unido por médicos del Centro de Fertilidad de Newcastle.
El proceso brevemente resumido consta de varios pasos: primero, el esperma del padre se usa para fecundar los óvulos de la madre afectada y una donante sana. Después se extrae el material genético nuclear del óvulo de la donante y se reemplaza con el del óvulo fertilizado de la pareja.
El óvulo resultante tiene un conjunto completo de cromosomas de ambos padres, pero lleva las mitocondrias sanas de la donante en lugar de las defectuosas de la madre. Después se implanta en el útero.
Mi pregunta es, ¿creéis que a partir de ahora gracias a técnicas como esta van a evitarse muchas enfermedades hereditarias?
Un equipo de investigadores de las universidades de Nueva York y Toronto han desarrollado ZFDesign; el primer método del mundo para editar proteínas basándose en la activación y desactivación de genes. Este gran avance promete ser más seguro y eficaz que el ya conocido CRISPR a la hora del tratamiento de enfermedades cardiovasculares, obesidad y autismo.
CRISPR es una herramienta de edición genética que permite tratar enfermedades provocadas por un error en la disposición de las bases del ADN. Hay otras enfermedades, las epigenéticas, que no tienen que ver con este error, sino que están causadas por la manera en la que las células leen esa información.
Los genes llevan los planos de ensamblaje de las proteínas y los factores de transcripción se encargan de decirle a la célula cuántas unidades tiene que fabricar. Pero en ocasiones se dan fallos de comunicación que hacen que esas cantidades se superen o no lleguen al número que se necesita, provocando enfermedades como la diabetes, el cáncer o trastornos neurológicos.
El nuevo sistema ZFDesign trata de solucionar este problema. Las nucleasas dedo de cinc (ZFN) emplean dominios de unión de ADN personalizables que se unen a dominios de escisión de ADN no específicos.
¿CÓMO FUNCIONA?
Según la noticia, cada dedo de ZFN contiene dos dominios distintos: una proteína dedo de cinc que comprende un DBD compuesto por dos componentes de “dedo” cosidos juntos y un dominio nucleasa de escisión de ADN. Ambos componentes combinados conforman una especie de tijera que permite la escisión de la doble cadena de ADN en un punto determinado, y a su vez estimulan la unión de extremos no homólogos propensos a este tipo de errores.
La diana de esta molécula son los dedos de cinc; presentes en los factores de transcripción, las proteínas de unión o las enzimas, y desarrollan importantes acciones como el control del funcionamiento de los genes y la interacción con otras proteínas.
Sin embargo, crear dedos de zinc que desarrollen su función de manera procesia no es tarea fácil. Estas proteínas se unen al ADN formando grupos complejos, lo que obliga a los investigadores a tener que seleccionar, entre las innumerables combinaciones posibles, la manera en la que cada dedo de zinc interactúa con los demás elementos para realizar cada uno de los cambios genéticos deseados.
Para solucionar este inconveniente, los investigadores se han servido de la inteligencia artificial. Así, han creado un modelo alimentado con datos de cribado de casi 50.000 millones de posibles interacciones entre dedos de zinc y ADN.
Para verificar la eficacia del modelo, los investigadores crearon un dedo de cinc específico que alterara la secuencia de codificación de un gen humano. Asimismo fabricaron otros que eran capaces de cambiar la forma en que los factores de transcripción se unían a una secuencia genética como la propia expresión del gen.
«Nuestro programa puede identificar la agrupación correcta de dedos de zinc para cualquier modificación, lo que hace que este tipo de edición de genes sea más rápida que nunca», afirma el doctor David Ichikawa, antiguo estudiante de posgrado en la Universidad de Nueva York y uno de los autores principales del estudio publicado recientemente en la revista Nature Biotechnology.
UNA HERRAMIENTA QUE SUPERA A CRISPR
El mismo doctor afirma que la edición genética con dedos de zinc puede ser una alternativa más segura que CRISPR, ya que esta última emplea proteínas bacterianas —ajenas al cuerpo humano y que pueden por tanto activar los sistemas de defensa inmunitarios de los pacientes al confundirlas con una infección—. Sin embargo los dedos de zinc sólo los fabrican los humanos y son mucho más pequeños, con lo que suponen un riesgo mucho menor para el sistema inmunitario.
A pesar de este prometedor avance, el equipo asegura que los dedos de zinc son difíciles de manejar. Algunas combinaciones no son exclusivas de un solo gen y pueden alterar secuencias de ADN que no son el objetivo inicial, provocando cambios no deseados en el código genético. Para solucionarlo, los investigadores están trabajando en mejorar el algoritmo de IA para hacer grupos más precisos de dedos de zinc que únicamente provoquen el cambio que se busca.
«Nuestro sistema allana el camino para utilizar estas proteínas para controlar múltiples genes al mismo tiempo», explica el doctor Marcus Noyes de la NYU, otro de los autores principales del estudio. «En el futuro, este enfoque podría ayudar a corregir enfermedades que tienen múltiples causas genéticas, como las cardiopatías, la obesidad y muchos casos de autismo».
REFERENCIAS
Gaj, T., Gersbach, C. A., & Barbas III, C. F. (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends in biotechnology, 31(7), 397-405.
1. J. Doudna and E.J. Sontheimer. “The Use of CRISPR/Cas9, ZFNs, and TALENs in Generating Site-Specific Genome Alterations,” Methods in Enzymology 2014.
Según lo publicado en la revista Nature a día 15 de marzo de 2023, un equipo de investigadores de la Universidad Kyushu (Japón), dirigido por Katsuhiko Hayashi, ha logrado obtener óvulos funcionales a partir de células madre pluripotentes derivadas de ratones macho y tras fecundarlos e implantarlos en ratones hembra, han generado embriones de ratón, que tras su desarrollo, han resultado en ratones adultos fértiles y aparentemente normales.
El trabajo presentado no es el primero en abordar la posibilidad de obtener embriones a partir de células de origen exclusivamente femenino o masculino, dado que en 2018 un grupo de investigadores de la Academia China de la Ciencia reprogramó el epigenoma de células madre embrionarias haploides de ratón que fusionaron a gametos para generar embriones cuyas células fueran todas de origen masculino o femenino. No obstante, en esta ocasión únicamente progresaron los embriones de origen femenino.
En el trabajo presentado en la III Cumbre Internacional de Edición del Genoma Humano, el equipo de Hayashi expuso que el experimento se llevó a cabo a partir de células madre pluripotentes inducidas (iPSC), es decir, células diferenciadas que sn reprogramadas a un estado no diferenciado con potencial para dar lugar a múltiples tipos celulares. Dichas células se obtuvieron mediante la inducción de la sobreexpresión de los factores de transcripción KLF4, SOX2, OCT4 y cMYC.
Una vez obtenidas las células madre pluripotentes inducidas, a partir de un ratón macho, las mantuvieron en cultivo hasta que de forma espontánea algunas de ellas perdieran el cromosoma Y. Posteriormente, el equipo indujo la duplicación del cromosoma X y generó ovocitos, que tras comprobar que no presentaban anomalías cromosómicas y que eran prácticamente idénticos a los generados directamente de células madre embrionarias femeninas, se procedió a la fecundación del ovocito y la posterior implantación del embrión en ratones hembra.
Finalmente, tras 630 embriones transferidos, los investigadores tan solo obtuvieron 7 ratones adultos fértiles, lo que se traduce en una tasa de éxito muy baja. Esto implica que que todavía se desconocen muchos de los mecanismos implicados.
A pesar de que se trata de resultados muy preliminares, existen diferentes aplicaciones potenciales. En primer lugar, cabría la posibilidad de restaurar la fertilidad en personas con alteraciones en los cromosomas sexuales, como sería el caso de aquellas con síndrome de Turner, en las que solamente existe un cromosoma X, mientras que para la producción de un ovocito se requieren dos. Por otra parte, también podría servir para investigar mutaciones recesivas ligadas al cromosoma X, así como para generar embriones en los que ambas copias de los cromosomas sexuales proceden del mismo progenitor.
En definitiva, el proyecto contribuye a conocer mejor los mecanismos implicados en el desarrollo y maduración de los gametos, lo que podría ayudar a conocer mejor la infertilidad. No obstante, su aplicación con finalidad reproductiva en humanos se halla muy lejos. Esto se debe a que el proceso estudiado en ratones difiere al de humanos en múltiples aspectos, entre los que destaca el tiempo de cultivo, que al ser mayor en humanos, contribuiría a un mayor riesgo de desarrollar alteraciones genéticas y epigenéticas. Además, en el caso de que se consiguiera trasladar la estrategia a un escenario humano, se precisaría de una gestación subrogada, entrando en juego la ética de su aplicación.
REFERENCIAS
Murakami, K., Hamazaki, N., Hamada, N. et al. Generation of functional oocytes from male mice in vitro. Nature (2023)
El desarrollo de la inmunoterapia y su impacto en el tratamiento del cáncer son el fruto de muchos años de investigación. La inmunoterapia, en particular, tiene como objetivo aprovechar, manipular y potenciar la defensa del propio organismo contra las células tumorales.
La inmunoterapia usada como antitumoral hoy en día es el principal sector en desarrollo de ensayos clínicos a nivel global. Englobando un amplio abanico de posibilidades terapéuticas, sin embargo, actualmente se estudia en gran medida las estrategias de inmunoterapia adoptiva con células T, basadas en modificaciones del receptor de estos linfocitos T y el desarrollo de receptores antigénicos quiméricos (CAR)
El receptor de antígeno quimérico es un receptor sintético, el cual se trata de una proteína encontrada en la membrana de la célula. Consta de un dominio extracelular, el cual se une al Ag tumoral específico, la región bisagra, el dominio transmembrana y el dominio intracelular que posee la cadena Z del complejo CD3 que activa a la célula T
Por definición, todas las células CAR-T, tanto autólogas como alogénicas, son linfocitos T modificados genéticamente para expresar un receptor CAR específico en su membrana.
La inmunoterapia CAR surge de la combinación de los avances en terapia humoral específica contra el tumor y la manipulación genética de los linfocitos T, formando las células CAR-T. Una célula CAR-T está compuesta de los dominios de reconocimiento antigénico obtenidos de las regiones variables de las cadenas pesada y ligera de un anticuerpo monoclonal específico, unidas entre sí para formar una anticuerpo de cadena simple (scFv).
Este anticuerpo está acoplado a un dominio de señalización derivado del dominio intracelular de la cadena CD3ζ del receptor de la célula T-CD3. Los primeros CAR-T formados de esta manera fueron nombrados de primera generación.
Hoy en día la técnica explicada anteriormente ha evolucionado para incorporar a la estructura de señalización distintos dominios procedentes de distintas moléculas consideradas coestimuladoras como por ejemplo CD28, PX40 o 41BB, dando lugar a los CAR-T de segunda y tercera generación.
Por lo que pueden diseñarse CAR-T contra cualquier antígeno extracelular. A destacar las células CAR-T dirigidas contra el antígeno CD19 (llamadas CART-19) expresado en linfomas y leucemias linfoblásticas agudas de tipo B representando el mayor éxito clínico por el momento.
En España en paralelo se desarrollan CART-19 en instituciones académicas
Esta noticia mencionada, que fue publicada el 9 de enero de 2023, está basada en un proyecto con participación del CSIC en el cual se desarrollarán biocápsulas capaces de transportar células antitumorales CAR-T para invadir la zona cancerosa y acabar con las células malignas desde el interior del tumor.
Se trata de un ambicioso proyecto basado en el uso de células inmunitarias diseñadas específicamente para atacar moléculas expresadas en algunos tipos de tumores, estas son las células CAR-T. El objetivo es usarlas para tratar de una manera más efectiva y específica tumores sólidos.
Las células CAR-T estarán incluidas en cápsulas, estas capsulas diminutas van a ocultar en su interior las células modificadas pudiendo invadir la zona cancerosa acabando con las células malignas.
En este proyecto van a participar el Centro de Investigación contra el Cáncer de Salamanca (CIC) que es un instituto mixto de investigación del CSIC y la Universidad de Salamanca. También va a participar el Centro de Investigación Biomédica en Red de Cáncer (CIBERONC), la Universidad de Santiago de Compostela (USC) y el Cima Universidad de Navarra.
Las terapias con el uso de las células CAR-T ya son usadas en tratamientos oncológicos pero con muy baja eficacia ante los tumores sólidos. En búsqueda de mejorar estas terapias las biocápsulas que van a desarrollar favorecerán la implantación y diseminación localizada de las células CAR-T en este tipo de tumores.
También se va a incluir dentro de las células diferentes mezclas de moléculas que faciliten la actividad de las células a largo plazo, como la modificación del microambiente tumoral para que este ambiente sea receptivo para la acción antitumoral de las células y la permita.
“Con esta metodología queremos emplear la táctica del caballo de Troya: introducir en el tumor células CAR-T escondidas en una cápsula protectora para que, una vez liberadas, provoquen la destrucción de las células tumorales que las rodean de la forma más efectiva posible”
“Para ello, añade, las biocápsulas incluirán cócteles de moléculas biológicas dirigidas a facilitar la acción de las células CAR-T liberadas dentro del tumor. Usando un símil bélico, es como poder bombardear toda una zona de guerra tras haber inactivado las defensas antiaéreas del tumor”
el investigador del CSIC Xosé Bustelo
La clave de este proyecto que nos llena de esperanza es el uso de las cápsulas como caballos de Troya que nos permita mantener a soldados que serían estas células modificadas dentro de ellas y facilitarles armas que permitan que estos soldados sean los más letales posibles contra las células tumorales.
Tal y como explica Sandra Hervás (CIMA) como una de las investigadoras principales del proyecto. Van a usar como modelo de trabajo diversos subtipos de cáncer de mama que serán tratados con células CAR-T modificados genéticamente para que reconozcan moléculas que se expresan de forma específica en cada uno de dichos subtipos. El objetivo principal del proyecto es que más adelante estos tratamientos sirvan con ligeras modificaciones para otros tipos de tumores sólidos.
Se ha descubierto que otras aplicaciones de estas cápsulas es incorporar materiales magnéticos, que con una estimulación externa, permitan destruir células tumorales generando altas temperaturas dentro del tumor. El proyecto contempla también el estudio del interés potencial del uso simultáneo de esta con la quimioterapia o terapias dirigidas.
Tras haberse contrastado a nivel experimental en modelos celulares y animales, esta versión de inmunoterapia va a ser probada en ensayos clínicos, en manos de la empresa pública gallega de servicios sanitarios GALARIA
“Con este abordaje, queremos cerrar en el proyecto todo el ciclo que va desde la innovación en el laboratorio hasta la implementación práctica a nivel clínico de estas terapias”
Alicia Piñeiro
Este innovador proyecto llamado como Encapsulación de células CAR-T en sistemas porosos nanoestructurados bioactivos para su liberación dirigida en tumores sólidos ha sido concedido por la Agencia Estatal de Investigación del Ministerio de Ciencia e Innovación.
PUNTOS DÉBILES DE LA TÉCNICA
Hoy en día se plantean problemas como el uso de estas células contra tumores sólidos, también el medioambiente desarrollado por las células tumorales que es hostil para estas células inmunitarias y promueve su inactivación a corto plazo, una disminución de su eficacia a largo plazo debido a la diseminación de las células CAR-T inyectadas por todo el cuerpo del paciente.
Por último otro inconveniente es que alguna de estas terapias pueden tener efectos colaterales debido a que las moleculas que son reconocidas por las células CAR-T están presentes en algunos casos en células de nuestro cuerpo que no están implicadas en la enfermedad pudiendo ocasionar otros daños en tejidos que no tienen relación con la enfermedad
BIBLIOGRAFÍA
Acebal Arranz, C. (2022). Estrategias de producción de células CAR-T para uso alogénico: una revisión sistemática.
Mirones, I., Moreno, L., Patiño-García, A., Lizeaga, G., Moraleda, J. M., Toribio, M. L., … & Menéndez, P. (2020, July). Inmunoterapia con células CAR-T en hematooncología pediátrica. In Anales de Pediatría (Vol. 93, No. 1, pp. 59-e1). Elsevier Doyma.
CAR, D. D. L. T. Inmunoterapia CAR-T: la nueva era en el tratamiento del cáncer Comparte este post.
La noticia que comentaré a continuación fue publicada por el Centro Nacional de Investigaciones Oncológicas (CNIO) el 2 de noviembre de 2022 y cuyo artículo científico se publicó en la revista científica estadounidense Science Advances (volume 8, issue 44).
Carolina Villarroya, investigadora del CNIO, junto con Marcos Malumbres, jefe del grupo de División Celular y Cáncer del CNIO
La protagonista de esta historia es una mujer española de 36 años que ha sufrido doce tumores a lo largo de su vida, siendo cinco de ellos malignos. No se sabía el porqué de estos hechos tan sorprendentes. Los científicos del CNIO afirmaban que un caso así era muy extraño y cuyas posibilidades de supervivencia eran ínfimas.
Para poder averiguar la causa de estos tumores se decidió realizar un estudio genético de esta propia mujer y de sus familiares (padres, hermanos, …). En un principio no se pudo llegar a una conclusión ya que todo resultó favorable en los árboles familiares. Por esta razón se quiso investigar aún más su caso a través de la secuenciación de células únicas.
Esquema tomado de ‘Biallelic germline mutations in MAD1L1 induce a syndrome of aneuploidy with high tumor susceptibility’, publicado en Science Advances vol.8 no,44 (DOI: 10.1126/sciadv.abq5914) A) Cromogramas que muestran las dos mutaciones heterocigotas en el gen MAD1L1 de la paciente; B) Representación esquemática de la proteína MAD1 y las mutaciones encontradas en la paciente; C) Estructura predicha de MAD1; D) Pedigree de la familia
Estudiando el genoma de miles de células de la afectada y sus familiares se pudo observar que la mujer no presentaba mutaciones en los genes que suelen desencadenar la formación de un tumor en caso de alteración (p53, BRCA1, BRCA2, PALB2, STK11). Sin embargo, se observó que presentaba una doble mutación en el gen MAD1L1, heredada una copia mutada por cada uno de sus progenitores.
El gen MAD1L1 se encuentra en el brazo corto del cromosoma 7, en la posición 22.3 (7p22.3). Este gen codifica para unas proteínas esenciales en el ciclo celular que intervienen en el punto de control del ensamblaje correcto de los microtúbulos en la anafase. Si los cromosomas no están bien alineados en esta fase del ciclo, este no continuará hasta que se solucione el problema. La afectada presentaba una mutación bialélica en este gen, produciendo en consecuencia un desbalance cromosómico en todas sus células (aneuploidías). La proteína MAD1 también se asocia con la proteína MAD2 en este proceso.
Imagen tomada de Genecards – Cromosoma 7 donde encontramos indicada en rojo la posición del gen MAD1L1Estructura proteica de MAD1 (en color verde) asociada con MAD2a (en color rosa) – Realizado con el programa informático ChimeraX (PDB 1GO4)
Este gen solamente se había estudiado en ratones hasta el momento, por lo que la información de este en humanos era todavía escasa, aunque se está investigando en los laboratorios a día de hoy. La afectada es el único caso humano que presenta esta doble mutación y los científicos no entendían cómo podía haber sobrevivido no a los tumores sino a los mecanismos de seguridad que presentamos en nuestro propio organismo, pudiendo no haber llegado a la vida a causa de un aborto natural. Por un lado, no se ha podido dar un nombre a esta enfermedad ya que solamente la padece ella; pero por el otro, se ha podido archivar como un síndrome de aneuploidía en mosaico variegadas (Mosaic Variegated Aneuplody syndrome – MVA), cuya prevalencia es de una persona por cada millón de habitantes. Ejemplos de genes que pueden causar este síndrome son BUB1B o CEP57, todos ellos también relacionados con el crecimiento celular.
Las características clínicas más frecuentes de los enfermos de MVA son microcefalia (tamaño de la cabeza más pequeño del esperado), micrognatia (mandíbula pequeña), anomalías del sistema nervioso central o anomalías oftalmológicas entre otras, como la mayor prevalencia de sufrir cáncer.
Imágenes tomadas del documento de materiales suplementarios de ‘Biallelic germline mutations in MAD1L1 induce a syndrome of aneuploidy with high tumor susceptibility’, publicado en Science Advances vol.8 no,44 (DOI: 10.1126/sciadv.abq5914)
Con el estudio se pudo llegar a la conclusión de que esta mutación bialélica perturbaba el crecimiento celular natural, haciendo que se vuelva precipitado, descontrolado y cuyas células presentaban aneuploidías a causa de errores en la segregación cromosómica.
La gran pregunta era: ¿cómo ha podido sobrevivir la paciente estos años teniendo en cuenta su condición médica y genética?
La respuesta se hizo esperar, pero gracias al estudio de su caso en concreto se pudo establecer una razón coherente. Su propia enfermedad le había estado protegiendo en todo momento. La inestabilidad genómica que presentaban alrededor del 40% de sus células sanguíneas desencadenó procesos inflamatorios muy frecuentemente, lo que indujo a su sistema inmunitario a estar alerta y muy estimulado en todo momento. Por esta razón, el hecho de la inestabilidad genómica facilitó la destrucción de los tumores por el sistema inmunitario tras las cirugías y tratamientos oncológicos, puesto que ellos también presentan esta inestabilidad.
Gracias al uso de la secuenciación de células únicas se pudo también detectar la presencia de pequeñas colonias celulares con defectos cromosómicos que son muy comunes en la leucemia, pudiendo así utilizar estas técnicas para la prevención del cáncer antes de que este comience a desarrollarse como un tumor. Sin embargo, en España no existe una especialidad de genética clínica en la que futuros investigadores puedan especializarse y poder estudiar a estos pacientes y no pasar por alto casos de cáncer hereditarios.
El gen p53 es un gen también llamado “guardián del genoma” debido a que codifica para una proteína supresora de tumores. En la especie humana se encuentra en el brazo corto del cromosoma 17. Su nombre se debe a su masa molecular aparente.
Cuando ocurre algún daño en el ADN esta proteína es esencial ya que induce la respuesta celular y detiene el ciclo celular en caso de mutación, es decir, p53 actúa principalmente como un factor de transcripción activado por estrés. Un p53 defectuoso podría permitir la proliferación de células anormales resultando en cáncer.
En condiciones sin estrés, p53 es ubiquitilado por MD,2 y otras ligasas E3 y posteriormente degradado por la maquinaria del proteasoma. Cuando existe une strés, como puede ser el daño del ADN, una infección vírica, activación oncogénica o el agotamiento de nutrientes, p53 se activa y se modifica postraduccionalmente.
Algunas de estas modificaciones consiguen evitar la degradación de p53 por parte de E3 y aumentar los niveles de estado estacionario de p53. La p53 activada se une a la cromatina y regula la transcripción de sus objetivos. Estas funciones celulares parecen contribuir a la supresión de tumores .
MUTACIONES EN P53
Cuando el gen p53 presenta alguna mutación no solo se altera su actividad antitumoral, sino que la mutación conferirá propiedades oncogénicas a la proteína p53. Numerosos factores estresantes endógenos y exógenos pueden activar p53 provocando que regule aún más respuestas celulares necesarias para mantener la homeostasis. Esta activación es fundamental para que las células normales sobrevivan y no se formen tumores.
Sin embargo, TP53 está mutado en la mayoría de cánceres, de manera que no puede realizar las funciones necesarias para suprimir el tumor y en ocasiones gana funciones que promueven el crecimiento de este. La mutación más común afecta solo a un aminoácido en la proteína p53 pero tiene un efecto significativo en su función.
Los tumores que albergan mutaciones de p53 normalmente suelen progresar de manera más rápida, tienen una respuesta deficiente frente a la terapia y suelen tener mal pronóstico. Por ello, dirigirse a este gen para la terapia contra el cáncer parece interesante.
DESARROLLO DE FÁRMACOS
Debido a su papel primordial en la supresión de tumores, p53 ha atraído un gran interés en el desarrollo de fármacos.
La terapia dirigida a este gen comenzó buscando compuestos que fueran capaces de restaurar o reactivar funciones de p53 salvaje (wtp53) o eliminar al p53 mutante (mutp53). El desarrollo de fármacos dirigidos a p53 es bastante difícil ya que el agente debe dirigirse específicamente a mutp53 en células cancerosas sin tener ningún efecto sobre las células con wtp53.
Nos vamos a centrar en las principales estrategias terapéuticas:
Recolocación del gen p53 de tipo salvaje wtp53 en las células cancerosas.
se busca realizarlo mediante terapia génica, es decir, la introducción de material genético en las células. Un estudio se centró en el desarrollo de un adenovirus recombinante que expresa wtp53 para tratar el carcinoma de cabeza y cuello.
Reactivación de funciones p53 suprimidas:
MDM2 es el principal regulador negativo de p53, que impide que este entre al núcleo y por tanto su unión al ADN y promueve la degradación proteasomal. A su vez, la expresión de MDM2 es inducida por p53 y por tanto forman un ciclo de retroalimentación negativa para controlar los niveles de estado estacionario de p53 en las células.
La amplificación y la sobreexpresión de MDM2 son mutuamente excluyentes con la mutación de p53, de manera que la sobreexpresión de MDM2 involucra p53 intacto en todos los tipos de cáncer.
Inhibir los MDM en cánceres con wtp53 parece ser una estrategia efectiva y se ha aplicado con éxito en diferentes investigaciones. Al inhibir MDM se inhibe el crecimiento tumoral ya que se reactiva p53 de tipo salvaje.
Hacer que p53 mutante vuelva a ser funcional:
Restaurar mutp53 terapéuticamente es más difícil que interrumpir la interacción entre MDM2 y p53. Las funciones principales de los reactivadores de p53 son:
Estabilización de la estructura de wtp53
Replegamiento o prevención del plegamiento incorrecto de mutp53
Restauración de la capacidad de unión al ADN de mutp53
Promoción de la expresión de mutp53 completo
El mecanismo principal y más estudiado de la acción de p53 es su unión a ADN que resulta en la activación o inhibición (menos frecuente) de la transcripción de los genes diana de p53. Esta función de unión al ADN está regulada por la interacción de p53 con una serie de proteínas nucleares. Por ello, se diseñaron reguladores de moléculas pequeñas para bloquear o promover tales interacciones.
CONCLUSIÓN
Aunque haya gran cantidad de estudios actualmente con buenas perspectivas sobre sus resultados, no se ha descubierto más que una pequeña parte. Los nuevos enfoques se basan en gran medida en la expresión de neoantígenos de mutantes de p53 en células tumorales.
El futuro de todas estas terapias parece ser positivo y la investigación y desarrollo de fármacos basados en este gen podrían estar cerca de la cura de muchos tipos de cáncer.
BIBLIOGRAFÍA:
Hu J, Cao J, Topatana W, Juengpanich S, Li S, Zhang B, Shen J, Cai L, Cai X, Chen M. Targeting mutant p53 for cancer therapy: direct and indirect strategies. J Hematol Oncol. 2021 Sep 28;14(1):157. doi: 10.1186/s13045-021-01169-0. PMID: 34583722; PMCID: PMC8480024.
Huang J. Current developments of targeting the p53 signaling pathway for cancer treatment. Pharmacol Ther. 2021 Apr;220:107720. doi: 10.1016/j.pharmthera.2020.107720. Epub 2020 Oct 29. PMID: 33130194; PMCID: PMC7969395.
Shen Y, White E. p53-dependent apoptosis pathways. Adv Cancer Res. 2001;82:55-84. doi: 10.1016/s0065-230x(01)82002-9. PMID: 11447765.
Guan YS, He Q, Zou Q. Status quo of p53 in the treatment of tumors. Anticancer Drugs. 2016 Oct;27(9):811-8. doi: 10.1097/CAD.0000000000000397. PMID: 27384591.
Investigadores del Instituto de Tecnología de Massachusetts han desarrollado PASTE, una nueva herramienta CRISPR, que permite insertar fragmentos de ADN de gran tamaño en posiciones específicas.
Pero primero, ¿En que consiste la herramienta CRISPR?
El sistema CRISPR-Cas9 clásico se basa en la utilización de una enzima Cas9 la cual corta ambas cadenas del ADN, y un ARN guía que posiciona a la enzima en el lugar especifico donde se quiere realizar el corte. Esta aproximación es muy útil cuando lo que se desea es inactivar un gen. Un inconveniente de Cas9 es que introduce un punto de rotura en el ADN que debe ser reparado por los mecanismos de reparación de la célula. Estos no son infalibles y durante el proceso se eliminan algunos nucleótidos, lo que puede llevar a inactivar el gen afectado. Otra posibilidad es añadir un ADN que actúe como molde para los mecanismos de reparación, en cuyo caso se puede corregir un error genético.
Figura 1: Mecanismo del sistema CRISPR-CAS9
Mediante la herramienta PASTE (de Adición Programable vía Elementos Diana Específicos de sitio, en sus siglas en inglés) se facilita la introducción de secuencias de hasta 36 kilobases (grandes fragmentos de ADN) en el genoma sin generar puntos de rotura en las dos cadenas de ADN. Una ventaja frente al CRISPR clásico es que puede ser utilizado en celulas en división y genera menos modificaciones en posiciones no deseadas.
De tal forma que se hace posible reemplazar genes ya que hasta el momento, la mayor parte de herramientas de edición estaban dirigidas a la inactivación de genes o corrección de mutaciones.
Una de las principales ventajas de esta herramienta es el corte e solo una cadena. Para ello utilizaron una enzima Cas9 (combinada con un ARN guía) que en lugar de cortar las dos cadenas del ADN solo corta una . Además se requería que tuviera la capacidad de cambiar un gen defectuoso por uno funcional, para ello recurrieron a un tipo de enzima que utilizan los virus para integrar su genoma en el de las bacterias: las serina integrasas.
Las serinas integrasas se encargan de reconocer secuencias especificas del genoma y facilitar la integración del ADN del virus. En la naturaleza hay más de 25.000 serina integrasas y por tanto con su correspondiente secuencia de destino. De tal forma que durante PASTE se detecta cual sería la combinación de serina integrasa y su respectiva secuencia de reconocimiento para poder editar el genoma.
Una vez obtenida la secuencia especifica necesaria, tiene que haber un elemento que la introdujera para las integrasas. Se utilizó una transcriptasa inversa o retrotranscriptasa unida a un fragmento de ARN complementario de esa respectiva secuencia.
Figura 2: Mecanismo de actuación: en primer lugar el ARN guía posiciona a Cas9 sobre la región de interés. Cas9 introduce un corte en una de las cadenas de ADN y a continuación, la enzima transcriptasa inversa, con ayuda del segundo ARN genera en ese punto una secuencia diana de integración para la serina integrasa. Finalmente, la serina integrasa introduce el fragmento de ADN que carga en el sitio de integración.
En cuanto a sus posibles usos, es una herramienta de gran interés en el reemplazamiento de genes defectuosos en la fibrosis quística, podría facilitar la inserción de genes de interés en terapias celulares…
Actualmente se están buscando nuevas formas de optimizar PASTE y evaluar su potencial en la investigación relacionada con las enfermedades genéticas.
Yarnall, M.T.N., Ioannidi, E.I., Schmitt-Ulms, C. et al. Drag-and-drop genome insertion of large sequences without double-strand DNA cleavage using CRISPR-directed integrases. Nat Biotechnol (2022). https://doi.org/10.1038/s41587-022-01527-4
El gen TP53es un gen supresor de tumores que codifica para la proteína p53, que tiene un papel fundamental en la división y proliferación celular, interrumpiendo el ciclo celular si la célula tiene el ADN dañado y así, evitar dar lugar a células cancerosas. Gracias a su relevante función, este gen ha recibido el sobrenombre de “guardián del genoma”. Previamente se conocía que este gen estaba directamente relacionado con la aparición de muchos tipos de cáncer, pero recientemente se ha descubierto que también está asociado con distintas patologías cardiovasculares.
Figura 1: Esquema del funcionamiento de la proteína expresada por el gen TP53
A lo largo del día, producimos millones de células sanguíneas, lo cual induce que algunas de ellas presentan mutaciones en su genoma. El fenómeno conocido como hematopoyesis clonal tiene lugar cuando una célula hematopoyética comienza a producir células con la misma mutación genética, lo que se asocia a enfermedades cardiovasculares. Ahora, se conoce que mutaciones en el gen TP53 aumentan el riesgo de padecer aterosclerosis, la causa de la mayor parte de enfermedades cardiovasculares y la principal causa de muerte en Estados Unidos.
Figura 2: Desarrollo de la aterosclerosis: se produce una acumulación de grasa, colesterol o células sanguíneas formando una placa que estrecha la arteria. Fuente: National Heart, Lung and Blood Institute, May 13 2022.
En el estudio realizado por el equipo de investigación del CNIC (Centro Nacional de Investigaciones Cardiovasculares), se analizó el exoma (conjunto de exones del genoma) de células sanguíneas de alrededor de 50.000 personas, y los resultados obtenidos concluyeron en que aquellas personas con mutaciones en genes TP53 tienen más posibilidades de padecer afecciones cardiovasculares.
Para corroborar esto, se realizó un estudio experimental con ratones. A un grupo de ratones se le trasplantaron células hematopoyéticas con la mutación en el gen TP53, y a las trece semanas se comparó con otro grupo de ratones no trasplantados, y se observó que, en los trasplantados, la aterosclerosis se había desarrollado mucho más rápidamente que en aquellos no trasplantados, debido a un crecimiento anormal de células del sistema inmune en la pared de las arterias.
Gracias a este estudio, se conoce que este gen, así como la hematopoyesis clonal contribuyen al desarrollo de enfermedades cardiovasculares. Este investigación, junto con otra realizada en 2021 en la que se demostró que mutaciones en el gen TET2 de células sanguíneas contribuye a la aparición de enfermedades cardiovasculares, serán de gran utilidad para proporcionar tratamientos y prevenir estas afecciones.
“En un futuro, esto podría utilizarse para diseñar estrategias de prevención personalizadas dirigidas a los efectos específicos de diferentes mutaciones”
Nuria Matesanz, investigadora del CNIC
Personalmente, me resulta muy curioso, y a la vez sorprendente, como un gen que se conocía su función desde hace años, ahora pueda ser la llave para abrir la puerta al tratamiento y prevención de una de las mayores causas de muerte del mundo: las afecciones cardiovasculares. Enfermedades como la cardiopatía isquémica, que es una de las principales causas de hospitalización de personas mayores de 65 años, podrían verse solucionadas gracias a estos estudios y así prolongar la esperanza de vida de personas de avanzada edad. Aun así, es necesario realizar más estudios para conocer más detalladamente el funcionamiento del ser humano y de este modo, proponer soluciones a problemas que hoy en día son difíciles de tratar y que en un futuro próximo esperemos que no lo sean.
Zekavat, S. M., Viana-Huete, V., Matesanz, N., Jorshery, S. D., Zuriaga, M. A., Uddin, M. M., Trinder, M., Paruchuri, K., Zorita, V., Ferrer-Pérez, A., Amorós-Pérez, M., Kunderfranco, P., Carriero, R., Greco, C. M., Aroca-Crevillen, A., Hidalgo, A., Damrauer, S. M., Ballantyne, C. M., Niroula, A., … Klarin, D. (2023). TP53-mediated clonal hematopoiesis confers increased risk for incident atherosclerotic disease. Nature Cardiovascular Research, 1–15. https://doi.org/10.1038/s44161-022-00206-6
Los telómeros son las regiones de ADN que corresponden a los extremos de los cromosomas. En células eucariotas, al poseer cromosomas lineales y una replicación semiconservativa, estos extremos se van acortando en la división celular en la mayoría de tejidos. Este proceso es reconocido como senescencia replicativa y es la causa del envejecimiento celular.
Sin embargo, hay algunas células que evitan el acortamiento de telómeros y por tanto consiguen la inmortalidad celular. Entre ellas encontramos células tumorales que son capaces de mantener la longitud de sus telómeros a través de su continua división celular. Un mecanismo conocido en melanomas para mantener la longitud de los telómeros es la activación de la telomerasa, enzima capaz de polimerizar ADN en telómeros.
Esquema simplificado del funcionamiento de la telomerasa. (Klug WS, Cummings MR, Spencer CA (2006) Conceptos de Genética. (8ªedición). Editorial Pearson Prentice Hall, Madrid.)
Para la activación de esta enzima, en un 75% de melanomas se produce la mutación en el promotor del gen TERT, el cual codifica para el componente catalítico de la telomerasa. De la misma manera, es conocido que esta mutación no es suficiente para mantener la longitud de los telómeros.
Por ello, un grupo de investigadores de la universidad de Pittsburg centró su estudio en las mutaciones adicionales requeridas para la inmortalización de estas células. El objetivo de la investigación era descifrar nuevos mecanismos del mantenimiento de los telómeros a partir de mutaciones en genes relacionados con los mismos.
Su estudio les llevó a analizar mutaciones en la región reguladora del gen ACD, la cual codifica para un componente del complejo shelterina, TPP1, implicado en la protección de telómeros y regulación de la actividad telomerasa. Al estar la mutación del gen ACD ubicada en regiones reguladoras del gen se crean más sitios de unión de factores de transcripción, con el resultado de una mayor expresión de TPP1.
Posteriormente, se comprobó la presencia de ambas mutaciones en melanomas cutáneos y resultó que estaban presentes en alrededor de un 5% de ellos.
(Chun-on et al. )
A través de cultivos celulares con diferentes variantes se ha demostrado que el aumento de TPP1 es clave para la reactivación de la telomerasa y el mantenimiento de la longitud telomérica.
En el gráfico se expone la longitud del telómero (eje de ordenadas) frente al tiempo transcurrido(eje de abscisas). Las líneas azules discontinuas muestran las variantes mutantes del promotor TERT y la línea roja continua representa el punto crítico de acortamiento. (Chun-on et al. )
Este gráfico obtenido sobre datos experimentales del estudio muestra que las variantes mutantes del promotor TERT retrasan el acortamiento de los telómeros pero no es suficiente para evitarlo por completo. De esta manera, los resultados del informe indican que las mutaciones en regiones promotoras de los genes ACD y TERT actúan de manera cooperativa para mantener la longitud de los telómeros y evitar la senescencia replicativa.
Por último, los investigadores dieron un paso más y observaron que las mutaciones en el promotor de TPP1 se daban principalmente en cáncer de piel, pero también se han reportado casos en otros tipos de cáncer. Este descubrimiento abre camino a nuevas líneas de investigación sobre el mecanismo de inmortalización de células tumorales y al desarrollo de posibles tratamientos contra el cáncer
Informe científico e imágenes: Chun-On, P., Hinchie, A. M., Beale, H. C., Gil Silva, A. A., Rush, E., Sander, C., Connelly, C. J., Seynnaeve, B. K. N., Kirkwood, J. M., Vaske, O. M., Greider, C. W., & Alder, J. K. (n.d.). TPP1 promoter mutations cooperate with TERT promoter mutations to lengthen telomeres in melanoma. https://www.science.org.
Somos un grupo de 4 profesores de la Facultad de Ciencias (Alberto Altés, Alfredo de Bustos, Juan Manuel González y Yolanda Loarce) que decidimos hace unos años explorar nuevas herramientas, enmarcadas en distintos proyectos de innovación docente, que fomentasen la participación en el proceso de enseñanza-aprendizaje y el interés por la Ciencia de nuestros alumnos.
Aunque el trabajo del profesor y de los estudiantes se ha incrementado considerablemente con la adaptación de los grados al Espacio Europeo de Educación Superior, hay cosas que han cambiado poco: el “rollo de las clases magistrales”, dónde todos nos encontramos cómodos, y la escasa motivación general para participar en las actividades voluntarias propuestas si “no entran en el examen”. Nos hemos empeñado en hacer atractivas nuestras asignaturas, pero somos conscientes de que tenemos que seguir buscando la manera de interesaros e implicaros en ellas, y de que empecéis a disfrutar y asombraros con lo que cada día nos ofrecen los avances en Biología.
El mes pasado, leí un artículo que trataba sobre el caso de Kathleen Forbbig. Aquí os lo explico:
En 2003, una mujer australiana, Kathleen Forbbig, fue condenada a 10 años de prisión por asfixiar y matar a sus cuatro hijos (2 niños y 2 niñas los cuales tuvo en ese orden). De hecho, es considerada como la peor asesina en serie del país. Los niños, que oscilaban entre los 19 días y los 18 meses, murieron repentina e inesperadamente mientras dormían. Sin embargo, en 2019 varios científicos trataron de dar una explicación a estas extrañas muertes súbitas en bebés, con la creencia de que podrían tener una base genética y con el objetivo de que se pueda reevaluar el caso y poder sacar de la cárcel a la mujer.
Actualmente, una investigación científica (en la que está involucrada la científica española Carola García Vinuesa y es apoyada por 2 premios Nobel) apunta a que los cuatro niños pudieron morir por causas naturales a causa de una mutación genética.
La científica española Carola García Vinuesa FOTO: UNIVERSIDAD NACIONAL AUSTRALIANA LA RAZÓN
En 2019, este equipo descubrió un gen mutante en las dos niñas, CALM2 G114R. Este gen también presente en la madre, se cree que fuera patógeno y causó la muerte de las dos niñas, Sarah y Laura. Los investigadores también informaron que los dos hijos varones, Caleb y Patrick, portaban dos variantes raras del genBSN, el cual se ha demostrado que causa epilepsia letal en ratones.
La calmodulina (CaM) es una proteína fijadora de calcio que depende de él y regula una serie de enzimas y canales iónicos, incluidos los esenciales para regular la contractilidad cardíaca. Específicamente, CaM es clave para la inactivación dependiente de calcio del canal de calcio Ca V1.2 de la membrana celular, y para el cierre oportuno del canal de liberación de calcio del retículo sarcoplásmico cardíaco, RyR2.
Las calmodulinopatías son síndromes de arritmia raros y potencialmente mortales causados por variantes patogénicas en uno de los tres genes (CALM1, CALM2 y CALM3) que codifican proteínas de calmodulina idénticas. Los principales hallazgos tras la investigación fueron que la madre y sus dos hijas eran portadoras de una variante novedosa y funcional en el gen CALM2 asociada con muerte súbita en la infancia y la niñez, y que se prevé que la variante sea arritmogénica.
La variante que se encuentra en las niñas (G114R) afecta al mismo aminoácido que una variante (idéntica salvo en ese aá) de una proteína codificada por CALM3 (G114W), que estaría implicada en la SCD (anemia drepanocítica o falciforme) de un niño y el paro cardíaco de otro. Las variantes G114R y G114W confirieron cambios estructurales similares en CaM. Ambas variantes afectan a la capacidad de CaM para detectar cambios en la concentración de Ca 2+ y perjudican la unión y regulación de los dos canales cardíacos de Ca 2+ Ca V1.2 y RyR2. En base a estudios previos y los datos presentados en este artículo, predijeron que las variantes G114R y G114W son patógenas y que, por lo tanto, los portadores son propensos a las arritmias cardíacas, que podrían causar insuficiencia cardíaca durante el sueño.
El primer y el segundo niño eran heterocigotos compuestos para variantes sin sentido raras en el gen BSN, cuya deficiencia se sabe que causa epilepsia letal de aparición temprana en ratones. Además, parece ser que BSN es un gen altamente intolerante a la pérdida de función, pero estas variantes requerirían de más investigación para determinar si explicarían la epilepsia severa y la ceguera del segundo niño.
Bibliografía: Brohus, M., Arsov, T., Wallace, D. A., Jensen, H. H., Nyegaard, M., Crotti, L., … & Schwartz, P. J. (2021). Infanticide vs. inherited cardiac arrhythmias. EP Europace, 23(3), 441-450.
En mi opinión, si no se han encontrado otro tipo de pruebas como signos de violencia, envenenamiento etc. debería reanalizarse el caso y sacar a la mujer de prisión ya que lleva 19 años allí y ha sido encarcelada sin pruebas y ahora hay una investigación científica que aporta evidencias que podrían explicar los sucesos de los 4 niños, pero sigue siendo un tema bastante delicado, ¿vosotros qué opináis?
Recientemente he visto un artículo publicado por el MIT que hablaba sobre la posibilidad de que las vacunas de ARN se administrasen vía oral mediante píldoras. El ARN se liberaría en el estómago y, de esa forma, se podría ofrecer una nueva manera de adminsitrar vacunas o terapias para enfermedades gastrointestinales.
Como todos sabemos, las vacunas se suelen administrar en forma de pinchazos para inyectar el ARN o el microorganismo, dependiendo de la vacuna. Sin embargo, hay personas que le temen a las agujas y rechazan las vacunas por ese motivo. Mediante la píldora, ese problema se podría solucionar. Además, no solo favorece la tolerancia ante las vacunas, sino que ese mecanismo se podría emplear para administrar otros tipos de ARN o ADN terapéuticos directamente al tracto digestivo, importante para enfermedades como las úlceras.
Llama la atención este nuevo método, ya que la función principal del aparato digestivo es precisamente la degradación de los alimentos que se toman vía oral. Asimismo, los ácidos nucleicos son especialmente vulnerables y sensibles a la degradación. La forma de proteger el ARN o ADN se encuentra en el desarrollo de una cápsula lo suficientemente resistente a los procesos de degradación digestiva.
En el artículo se comenta el diseño de la cápsula: está diseñada con una cúpula, la cual se orienta para que el medicamento contenido se pueda inyectar en el estómago. En los experimentos llevados a cabo con ratones, las proteínas codificadas en ese ARN se mostraban tanto en el estómago como en otros tejidos como el hígado. Sin embargo, en cerdos solo se localizaba en el estómago.
Imagen del artículo que muestra el modo de acción de la píldora diseñada
En un futuro se espera aumentar la captación de ARN en otros órganos cambiando la composición o administrando dosis mayores. Bajo mi punto de vista, esta línea de investigación es prometedora y muy interesante y plantea muchas posibilidades nuevas. No solo se puede ayudar a que las personas con miedo a las agujas sean más receptivas a las vacunas (y, por consiguiente, tener a todavía más población protegida frente a ciertas enfermedades), sino que también se podrían explorar nuevos tratamientos frente a otras enfermedades, como las úlceras anteriormente mencionadas. Además, puede ser una vía más directa para tratar enfermedades o infecciones y acelerar el tratamiento.
Como es sabido, en los seres humanos el sexo femenino es determinado cromosómicamente, siendo el homogamético. Por ello, las hembras serán XX.
En ocasiones, ocurren cambios cromosómicos numéricos, más concretamente, aneuploidía. Estas aneuploidias son más comunes en plantas; sin embargo, en humanos se han detectado monosomías y trisomías. Estas trisomías pueden ser en cromosomas autosómicos, dando lugar a enfermedades como el síndrome de Down o en cromosomas sexuales, dando lugar a mujeres con XXX, vulgarmente conocidas como “las superhembras”.
El síndrome triple X es una anomalía cromosómica relativamente frecuente. Sin embargo, no suele identificarse en el nacimiento al no presentar un fenotipo característico. Normalmente, el diagnóstico se realiza cuando la mujer presenta una insuficiencia ovárica primaria. Sin embargo, en ocasiones, provoca retraso mental tanto profundo como ligero, malformaciones en la cara y aparato genitourinario o cardiopatías.
En todas las mujeres, siempre se inactiva por azar uno de los dos cromosomas X. Esto ocurre aleatoriamente de forma temprana durante la biogénesis y es controlada por el centro de inactivación cromosoma X. Por ello, sólo un cromosoma X de cada célula está activo genéticamente y el otro está inactivo. Sin embargo, aproximadamente un 5-10% de genes del cromosoma X que se debieron inactivar, no lo hacen. Además, las regiones psudoautosómicas del cromosoma X no se inactivan.
En caso de contar con la trisomía X, se inactivan dos de los tres cromosomas. Pero las partes no inactivadas del cromosoma X son expresados por los tres cromosomas X y, por ello, hay una sobrexpresión de genes.
Aún hay muchas cuestiones no resueltas en la trisomía X, por ello, es necesario seguir investigando sobre la fisiopatología y el mecanismo genético involucrado en los problemas médicos asociados.
El pasado mes de junio se cumplieron 40 años de los primeros casos de infección oficial causada por el VIH. A lo largo de todo este tiempo esta infección que genera un síndrome de inmunodeficiencia adquirida se ha cobrado la vida de mas de 30 millones de personas.
En la actualidad se ha avanzado bastante en los tratamientos antivirales frente al VIH hasta conseguir incluso su control en gran parte de los pacientes. Sin embargo no se ha logrado desarrollar una vacuna que proteja contra la enfermedad como si se ha obtenido para muchas otras enfermedades infecciosas.
A día de hoy se están realizando ensayos en diferentes animales con un prototipo de vacuna cuya tecnología se basa en lo mismo que dos de las vacunas para COVID-19.
Primeramente ¿Qué es el VIH?
El VIH pertenece a la familia de los lentivirus y se clasifica en dos tipos: VIH-1 y VIH-2 que tienen un 40-50% de homología genética y cuentan con una organización genómica similar. Tanto el VIH-1 como el VIH-2 provienen de diferentes saltos inter-especie de virus que infectan en la naturaleza
Al igual que en todos los virus envueltos, la envoltura consiste en una bicapa lipídica,en esta envoltura se encuentran presentes algunas proteínas de la célula huésped y muy significativamente Env, la glicoproteína de envoltura del VIH. Env se encuentra anclada en la membrana y consiste en un hetero-trímero formado por tres moléculas llamadas glicoproteína 120 (gp120).
La dificultad de neutralizar la infección por VIH-1 reside en la proteína Env; ya que cuenta con gran variabilidad de la envoltura con 5 regiones hipervariables además de un alto nivel de glicosilación de Env impidiendo la unión de anticuerpos y por ultimo enmascaramiento conformacional, término que describe que el sitio de unión con los co-receptores (CCR5 ó CXCR4), no existe hasta que se organiza espacialmente después del cambio en la conformación y es por tanto muy poco susceptible a la neutralización mediada por anticuerpos
Y por otro lado el gen gag codifica las principales proteínas estructurales
Al igual que con las vacunas contra el covid-19, la vacuna experimental contra el VIH envía instrucciones a las células, para producir dos proteínas clave del virus del VIH (Env y Gag).
“Esta vacuna experimental de ARN mensajero combinas múltiples características que podrían superar las limitaciones de otras vacunas experimentales para el VIH por lo que representa una aproximación prometedora”, ha señalado Anthony Fauci, Director del del Instituto Nacional de Alergias y Enfermedades Infecciosas, institución que junto a la empresa Moderna Inc (empresa responsable de una de las vacunas de ARN para COVID-19) y otros centros ha liderado la investigación.
El funcionamiento de la vacuna se rige por nanopartículas lipídicas que contienen en su interior moléculas de ARN mensajero de dos proteínas virales.
Una vez en el interior de las células este ARN mensajero inoculado por medio de la vacuna se traduce en las dos proteínas del virus formando así partículas virales no infecciosas que permiten una respuesta optima del sistema inmunitario.
Los resultados en ratones tras dos inyecciones de la vacuna mostraron anticuerpos neutralizantes en todos ellos (prueba de la respuesta inmunitaria) y por tanto se decidió pasar a una similar del ser humano, los primates. Primeramente recibieron una única dosis y se fueron administrando dosis posteriores a lo largo del año, sin mayores complicaciones salvo la pérdida de apetito de los macacos.
Tras 14 meses desde la primera dosis todos los individuos vacunados habían alcanzado unos niveles de anticuerpos neutralizantes bastante altos y dirigidos hacia la gran mayoría de cepas del VIH. Además de los niveles de anticuerpo, fortaleció la actividad de las células T de los monos contra este virus infeccioso que son claves en la respuesta inmunitaria. Al ser expuestos semanalmente a SHIV, 2 de cada siete no estaban infectados por lo que los investigadores concluyeron:
»No es un resultado rotundo, pero muestra que el enfoque podría funcionar en principio»
Por lo que este proyecto ofrece un punto de partida prometedor para un desarrollo exitoso de vacunas contra el VIH
“Estamos refinando nuestro protocolo de la vacuna para mejorar la calidad y la cantidad de partículas víricas producidas”, ha señalado Paolo Lusso. “Esto podría aumentar más la eficacia de la vacuna y reducir el número de inoculaciones primarias y de recuerdo necesarias para producir una respuesta inmunitaria robusta”, indica el investigador.
Una vez confirmada la seguridad y eficacia de la vacuna, el siguiente paso que quieren llevar a cabo es un ensayo en fase 1 con voluntarios humanos.
La hemofilia es una enfermedad de origen genético conocida desde hace siglos. Está ligada al sexo, concretamente al cromosoma X, la cual genera trastornos en la coagulación sanguínea. Esto hace que aumente la tendencia del paciente a sufrir hemorragias. Existen tres tipos de hemofilia: A, B y C. Nos centraremos en la hemofilia A ya que es la más frecuente entre la población, con una incidencia de 1/5000 niños varones. Este tipo de hemofilia representa el 80 % de los casos diagnosticados, siendo la patología ligada al cromosoma X más frecuente.
La causa de esta enfermedad se debe a una mutación en el gen F8 (localizado en el cromosoma X), el cual se codifica para la proteína del factor de coagulación VIII. Con esta mutación puede alterarse el factor o reducirse a una cantidad inferior a la requerida por el organismo. Por lo general los pacientes que sufren esta enfermedad tienen antecedentes familiares aunque también pueden darse mutaciones de novo.
La hemofilia A presenta un patrón hereditario recesivo ligado al sexo, donde los factores de coagulación VIII y IX se encuantran alterados ocasionando déficit cuantitativo y funcional en la formación de coágulos sanguíneos. Tan solo se presenta en los hombres, mientras que las mujeres generalmente son portadoras aunque pueden manfiestar la enfermedad en casos de inactivación del cromosoma X. Debido al estado homocigoto del cromosoma X de los hombres, estos sufren la enfermedad; sin embargo, en las mujeres su condición heterocigota, hace que sean portadoras asintomáticas.
La inactivación del cromosoma X en mujeres sintomáticas y mujeres hemofílicas puede verse afectado por algunos mecanismos genéticos que desencadenan efectos relevantes en las mujeres portadoras. Algunas mujeres portadoras pueden ser sintomáticas y presentar episodios hemorrágicos en algunas ocasiones, como hematomas. Estos se encuentran directamente relacionados con los niveles en el plasma sanguíneo de factor VIII (hemofilia A) o IX (hemofilia B).
Para detectar la enfermedad se empelan diversas técnicas de laboratorio que miden los niveles normales y alterados de actividad plasmática de los factores de coagulación VIII y IX. El síntoma más comun es la hemorragia, cuya gravedad depende del nivel del factor VIII presente en el plasma. Generalmente estos episodios se dan a consecuencia de traumas en articulaciones (hemartrosis) , músculos y zonas vinculadas al SNC, generando lesiones en vasos sanguíneos. Otro tipo de hemorragias comunes incluyen: SNC, sistema gastrointestinal, sistema genital-urinario, mucosas nasal y hematomas en vías respiratorias.
Hemorragias típicas en hemofilia
Los principales tratamientos se enfocan en prevenir la hemorragia y revertir sus efectos, ya que esta enfermedad por el momento no tiene cura definitiva. Algunos son tratamientos profilácticos primarios a edades tempranas. Otros consisten en reposición a niveles adecuados del factor de coagulación deficitario en función del tipo de hemofilia, generalmente de manera intravenosa. También es necesario el uso de medicamentos como DDAVP (acetato de desmopresina) que estimula la liberación del F8 y ácido épsilon aminocaproico que previne que los coágulos se deshagan.
como os he comentado en clase, aquí os dejo un enlace al artículo que ha salido publicado en Nature la semana pasada en el que se habla de como puede evolucionar el virus. Son opiniones muy interesantes de biólogos expertos en evolución de virus, en las cuales se describe los posibles escenarios de evolución del SARS COV-2 en función de estudios realizados con otros virus que han producido infecciones en la población humana y también de conocimientos teóricos sobre mutación y aspectos relacionados, lo cual está muy en la línea con lo que estamos tratando ahora en clase, las mutaciones y su relación con la evolución.
Por otro lado, os animo de nuevo a que participéis en el Blog, bien con comentarios a las noticias que han subido otros compañeros, todas muy interesantes (algunas también relacionadas con la nueva variante Omicron y como ha podido surgir) o con aportaciones nuevas que consideréis apropiadas. Os recuerdo que no se trata de copiar y pegar noticias sin más, tenéis que intentar aportar opiniones propias, aunque no seáis expertos en la materia, se trata de establecer debates que puedan enriquecer vuestro conocimiento.
El síndrome de Rubinstein-Taybi (nombrado así en reconocimiento a los científicos que lo descubrieron) es una enfermedad rara producida por una mutación en los genes 16 y 22. Se considera enfermedad rara porque afecta a 1 de cada 100.000 nacimientos, tanto en hombres como en mujeres.
Dicho síndrome se da por un trastorno genético normalmente esporádico, es decir, único en la familia, producido por las deleciones de distintas regiones en los cromosomas 16 (región 16p13) y 22. Posteriormente se identificaron dos genes que actuaban conjuntamente favoreciendo la aparición del SRT, el gen CBP (en el brazo corto del cromosoma 16) y el gen EP300 (en el cromosoma 22).
El gen CPB codifica para la proteína de unión a CREB, la cual se activa mediante la fosforilación de sus residuos serina, que alteran la conformación de su dominio de transactivación, incrementando así su interacción con la maquinaria transcripcional.
Al activarse la proteína CREB, esta se une a secuencias de DNA llamadas “elementos de respuesta a AMPc” que incrementan o reducen la transcripción. Esto ocurre ya que el AMP cíclico es un segundo mensajero que regula la actividad de las quinasas (proteínas que regulan los mecanismos de señalización celular). A parte de en la transcripción, la proteína CREB participa también en la regulación hormonal del metabolismo glucídico, como respuesta al glucagón.
Sin embargo, las mutaciones producidas en el gen EP300 solo causan un pequeño porcentaje de los casos de este síndrome. Este gen codifica para la proteína p300, la cual regula la actividad de muchos genes, de diversos tejidos, en procesos de crecimiento y división celular, es crítica para el desarrollo normal antes y después del nacimiento y las mutaciones que afectan a dicho gen provocan que se inactive una de las dos copias del mismo en cada célula, interfiriendo en el desarrollo normal de esta.
Los síntomas más característicos son baja estatura, discapacidad intelectual en diversos niveles, anomalías oculares, defectos cardíacos y renales, problemas dentales, obesidad, microcefalia, tendencia a desarrollo de ciertos tumores (recientemente, se ha demostrado que la expresión de CREB se relaciona directamente con el potencial metastásico de melanomas murinos), leucemia…
Pero sin duda uno de los rasgos físicos más apreciables, y el cual da el apodo a este síndrome, son los pulgares y primeros dedos de los pies más anchos de lo normal, de ahí que el logo representante de esta enfermedad rara sea:
Referencias:
Tríptico de información proporcionado por la AESRT (Asociación Española del Síndrome de Rubinstein Taybi).
Ya eran conocidos los riesgos que conlleva un exceso en la ingesta de estas grasa saturadas en el aumento del colesterol, la obesidad o la aparición de enfermedades cardiovasculares. Incluso en 2017, el Instituto de Investigación Biomédica de Barcelona (IRB) evidenció la relación que existia entre una dieta rica en este aceite y el peligro de desarrollar una metástasis del cáncer.
En este ya detallamos el proceso, desvelamos que existe un factor de ‘memoria’ de la capacidad metastática y señalamos una vía terapéutica para revertirlo”, subraya Aznar-Benitah.
Un nuevo estudio publicado en la revista Nature indica cómo una dieta rica en ácido palmítico (componente principal del aceite de palma), altera el genoma del cáncer y aumenta la probabilidad de que se extienda. Esto se debe a que las modificaciones que genera el ácido graso sobre el genoma de las células metastáticas son permanentes.
Los científicos vieron que las modificaciones que generan el ácido graso sobre el genoma de las células metastáticas pueden perdurar en el tiempo durante meses incluso cuando el ácido palmítico se había eliminado por completo de la dieta. Y concluyeron describiendo este comportamiento como una forma de memoria celular. Esta memoria es causada por cambios en el funcionamiento de los genes que modifican la función de las células cancerosas metastásicas y les permiten crear una red neuronal alrededor del tumor para comunicarse con las células de su entorno inmediato y diseminarse más fácilmente. Los distintos resultados apuntan a que la agresividad no solo tiene que ver con el metabolismo de las grasas, sino también con las modificaciones epigenéticas que se producen en las células tumorales cuando ingieren ácido palmítico. Para que una célula tumoral pueda establecer una metástasis, primero debe desprenderse del tumor de origen, introducirse en los vasos sanguíneos o linfáticos, alcanzar otro órgano vital y sobrevivir y crecer allí.
“Este descubrimiento abre nuevos caminos para la investigación y el desarrollo de terapias dirigidas a evitar específicamente la metástasis del cáncer que es, casi siempre, donde reside la mortalidad”, concluye la Dra. Gloria Pascual, investigadora asociada del laboratorio de Células Madre y Cáncer del IRB Barcelona y coprimera autora del artículo junto a la Dra. Diana Domínguez.
Esto nos lleva a pensar que en un futuro, cuando estos estudios sean comprobados en humanos y tengan eficacia, podriamos llegar a coseguir frenar la metastasis; un gran avance para la ciencia y una mejoria en la salud de la poblacion
Desde el 26 de noviembre de 2021, una nueva variante del SARS-CoV-2 ha sido reconocida por la OMS y denominada con la letra griega ómicron. La presencia de mutaciones que la diferencian de las otras cepas existentes y la gran cantidad de estas, así como el aumento de casos producidos en regiones concretas, han sido los factores determinantes para su denominación como variante preocupante. Esta ha sido notificada por primera vez en Sudáfrica y actualmente se están llevando a cabo numerosos estudios para averiguar más sobre ella; de momento se ha averiguado que el riesgo de reinfección es mayor respecto a otras cepas, aunque no parece que los síntomas sean muy graves, pero aún queda mucho que investigar.
Entre las más de 50 mutaciones presentes en ómicron la
mayoría se encuentran proteína de espiga y el dominio de unión
del receptor, dos zonas que intervienen en la adhesión y entrada del
patógeno a las células. Pero lo realmente destacable de esta variante a nivel genético
es el gran número de mutaciones que se han podido acumular, de forma aleatoria
pueden ocurrir mutaciones puntuales en la replicación del virus, pero en este
caso encontramos una acumulación de mutaciones que ya habían sido observadas en
otras cepas. Esto ocurre por el fenómeno de recombinación viral; dos
cepas distintas de un mismo virus pueden infectar una misma célula de manera
simultánea, y al replicarse el material genético de estos virus en su interior
pueden dar lugar a una nueva variante híbrida que compartirá mutaciones de
ambos.
En el caso de este virus, ha habido varios factores que han contribuido a que un hecho que parece tan poco probable haya sucedido. En primer lugar, el virus del Covid-19 posee ARN como material genético, y este es inestable y no posee mecanismos de revisión de su secuencia molecular durante la replicación, lo que provoca que haya un mayor número de mutaciones puntuales que en el caso del ADN. Por otra parte, el hecho de que este sea un virus tan extendido en la actualidad y haya un gran número de variantes que pueden convivir en una misma región es lo que ha propiciado una coinfección que finalmente ha dado lugar a esta recombinación.
Además, aunque este es el principal caso conocido de variante de SARS-CoV-2 que se ha originado por este mecanismo, no es el primero, ya que en febrero de este mismo año ya se identificó una muestra de virus que había sido recombinada a partir de la variante B.1.1.7 detectada inicialmente en Kent (Reino Unido) y la cepa B.1.429 originada en el sur de California, aunque solo fue identificada en laboratorio y no se detectaron casos en la población general.
La reciente variante Ómicron del Sars-Cov-2, actualmente conocido como coronavirus, ya se empieza a encontrar por todo el mundo. Descubierta en África, ya hay casos confirmados en España y recientemente en Canadá. Científicos no han perdido tiempo y directamente se han puesto a analizarlo, descubriendo que cuenta con un número inusualmente alto de mutaciones que le confieren al coronavirus el potencial de ser más transmisible y menos susceptible a las vacunas existentes. Sin embargo, todavía se desconoce mucho de esta nueva variante.
Investigadores han podido encontrar en la nueva variante más de 30 mutaciones en la proteína de la espícula. Dicha proteína es un componente que se encuentra en la superficie del vitus y que permite unirse a las células humanas y entrar de esa forma en nuestro organismo. Algunas muestras, sin embargo, han mostrado que había 50 mutaciones en todo el virus que hasta ahora no se habían encontraro juntas.
Las mutaciones ocurren de manera espontánea durante la reproducción del virus en el cuerpo de las personas. Al ser espontáneas, hay mutaciones que puede que no le confieran ninguna ventaja al virus, aunque hay mutaciones que pueden beneficiarle, al igual que puede suceder que le inhabiliten. Sin embargo, la selección natural y las medidas sanitarias acaban con los virus más débiles, mientras que los mejor adaptados pueden llegar a sobrevivir. La pregunta que surge es, ¿y cómo ha podido sumar todas esas mutaciones beneficiosas para el virus en una sola variante?
Antes que nada, es importante mencionar que, hasta que no terminemos con la pandemia, seguirán produciéndose nuevas mutaciones y variantes. Este proceso es algo inevitable si el virus se encuentra libre. Por ello, cuanto más tiempo se necesite en tener a la población vacunada (a nivel internacional, ya que vivimos en un mundo globalizado) y menos se respeten las medidas sanitarias como llevar mascarilla o mantener el espacio interpersonal, más difícil será detener la pandemia que ha paralizado el mundo por la aparición de más variantes.
Las variantes mejor adaptadas serán las que más se puedan reproducir y más contagiarán. Eso deriva de la ley de supervivencia, en el que el más fuerte logrará sobrevivir. Sin embargo, al confluir varias variantes, puede darse que, además de mutar las distintas cepas del virus cada una por su cuenta, también logren recombinar entre ellas. Puede darse el caso de que dos cepas mutadas con adaptaciones que les provean de ventajas, recombinen y formen una cepa nueva con ambas mutaciones y, por ello, ambos beneficios. De esa forma, y poco a poco, podría darse que una cepa logre sumar todas las mutaciones anteriores en un solo virus. Aunque estadísticamente puede parecer relativamente improbable, cuanto más extendido esté el virus, más puede entrar en contacto con distintas mutaciones, por lo que es importante frenar la expansión.
Según he leído en diversos artículos, la recombinación en los coronavirus, de forma común, se debe a la forma de replicación del genoma. La enzima que replica el material hereditario puede salirse de la cadena de ADN que está replicando y luego volver a unirse. De esa forma, en el momento en el que una célula huésped contenga los genomas de varias cepas distintas de coronavirus, la enzima puede saltar de un genoma al otro. Así se combinarían los genomas y se formaría un virus híbrido fruto de la infección mixta.
Otro tema preocupante con respecto a esta nueva variante es la eficacia de las vacunas. Expertos esperan que las vacunas proporcionen una cierta protección a la variante ómicron. Esto puede deberse a que, si las vacunas reconocen una cierta mutación en el virus, y la variante ómicron reúne muchas de las mutaciones que se han ido dando a lo largo de la pandemia, las vacunas reconozcan esa misma mutación en la nueva variante. No obstante, se están realizando experimentos y controles que denotan que la protección de las vacunas comienza a descender a partir de los seis meses de haber recibido la pauta completa. Por lo tanto, no estamos solamente expuestos a las mutaciones que se dan continuamente en el virus, sino también a la descendiente protección.
Aunque se intenten tomar medidas como confinamientos y dosis de refuerzo de las vacunas, la recombinación y la variabilidad del Sars-Cov-2 es inevitable. A la conclusión a la que he llegado es que, a pesar de ello, cuantos más impedimentos pongamos a frenar la pandemia, más recombinación va a darse y más trabas pondremos como sociedad a la ciencia. El buen trabajo de haber sacado las vacunas en poco tiempo se destroza con el cansancio de la población y de los movimientos negacionistas de parte de ella, los cuales solo favorecen la recombinación en el virus y la aparición de nuevas cepas más peligrosas.














































 Somos un grupo de 4 profesores de la Facultad de Ciencias (Alberto Altés, Alfredo de Bustos, Juan Manuel González y Yolanda Loarce) que decidimos hace unos años explorar nuevas herramientas, enmarcadas en distintos proyectos de innovación docente, que fomentasen la participación en el proceso de enseñanza-aprendizaje y el interés por la Ciencia de nuestros alumnos.
Somos un grupo de 4 profesores de la Facultad de Ciencias (Alberto Altés, Alfredo de Bustos, Juan Manuel González y Yolanda Loarce) que decidimos hace unos años explorar nuevas herramientas, enmarcadas en distintos proyectos de innovación docente, que fomentasen la participación en el proceso de enseñanza-aprendizaje y el interés por la Ciencia de nuestros alumnos.:quality(65)/cloudfront-eu-central-1.images.arcpublishing.com/larazon/3WIUZVGNCZFPBKVMJBMBPEEVXE.webp)

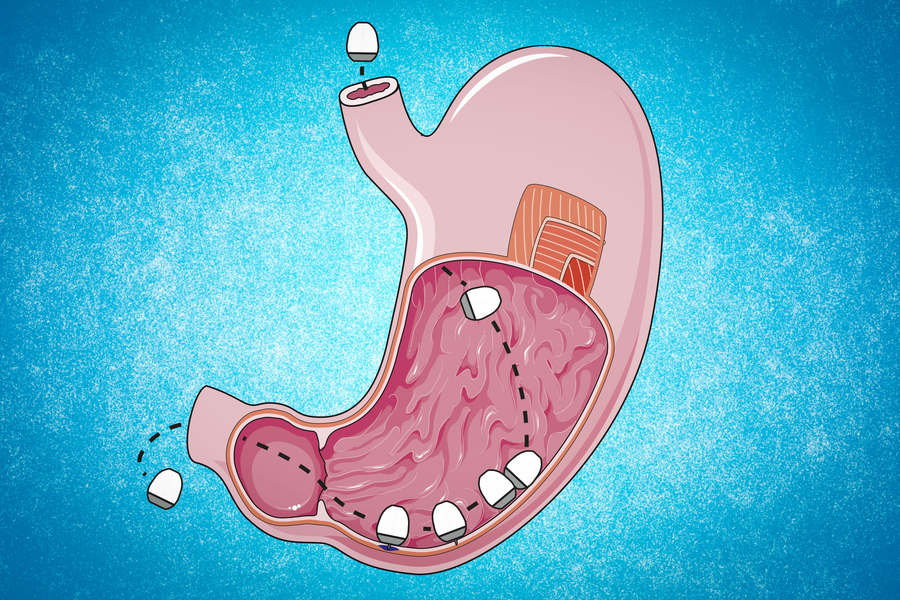
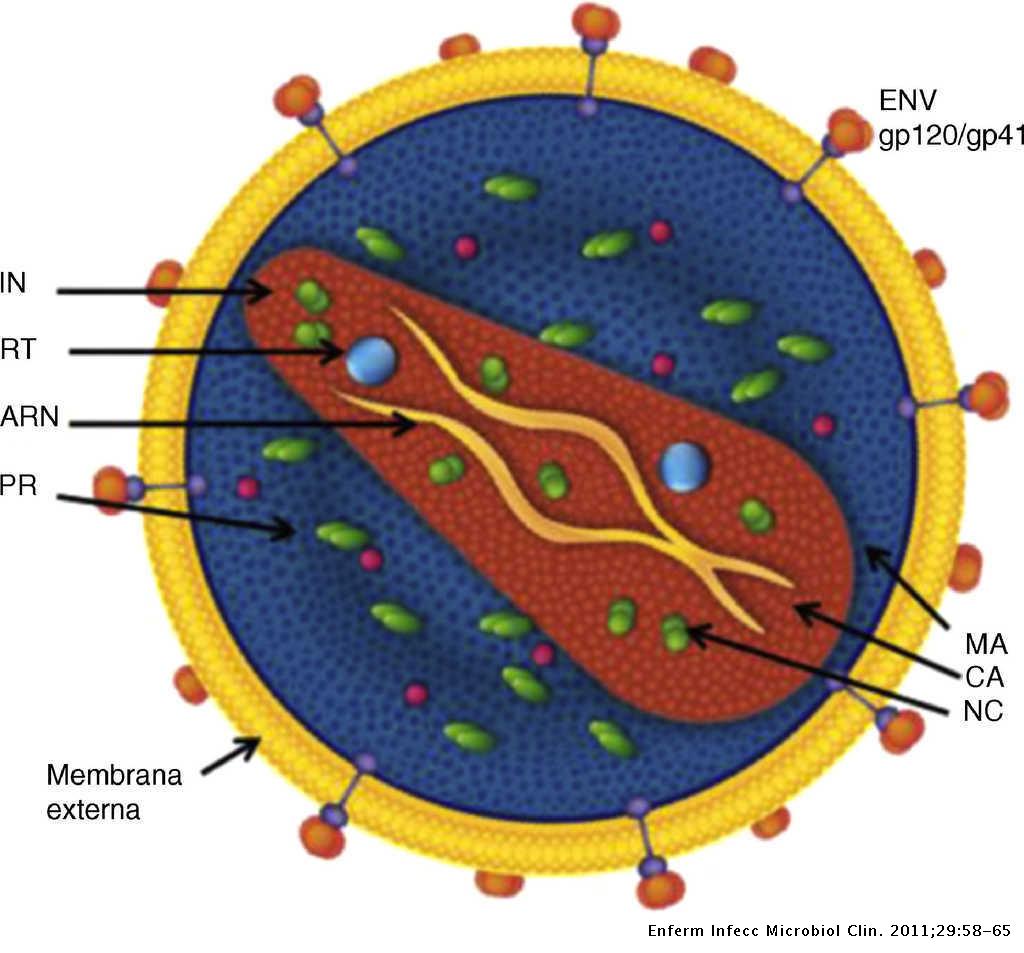





Debe estar conectado para enviar un comentario.